Product
Minimal amount stored
Acetaminophen/paracetamol 10 suppositories 600 mg
40
Acetaminophen/paracetamol 16 Tbl. 500 mg
400
Acetaminophen/paracetamol infusion solution 1 g 100 ml
9,600
Acetaminophen/paracetamol infusion solution 500 mg 50 ml
1,800
Albumin infusion solution 20 % 5 × 50 ml
10
Amoxicillin/clavulanic acid 1 g Ad 20 Tbl
180
Amoxicillin/clavulanic acid infusion solution 1.2 g 5 Ad Amp
750
Amoxicillin/clavulanic acid infusion solution 2.2 g 5 Ad Amp
350
Atracurium besylate injection solution 5 Amp 2.5 ml
100
Basic infusion solution G5-K PP 500 ml
600
Bupivacaine injection solution 0.25 % 5 Amp 20 ml
50
Bupivacaine injection solution 0.5 % 5 Amp 20 ml
30
Ceftazidime vial 2 g
250
Ceftriaxone 2 g vial
800
Cefuroxim injection solution 1.5 g i.v. vials
2,800
Ciprofloxacin infusion solution 0.2 g 100 ml
400
Clarithromycin injection solution 500 mg i.v. Amp
150
Clindamycin injection solution 600 mg 3 Amp
100
Desflurane 6 bottles 240 ml
8
Dihydralazine mesylate 25 mg 5 Amp
25
Diphtheria tetanus toxoid combination pre-filled syringe
300
Dobutamine concentrate infusion solution 250 mg
180
Epinephrine/adrenalin injection solution 1 mg/ml 10 Amp 1 ml
150
Epinephrine/adrenalin injection solution 1 mg/ml 10 Amp 10 ml
20
Etomidate injection solution 10 ml 10 Amp
40
Fentanyl injection solution 0.05 mg/ml 10 Amp 2 ml
300
Fentanyl injection solution 0.05 mg/ml 5 Amp 10 ml
600
Flucloxacillin injection solution 1 g 10 vials
150
Glucose 5 % NaCl 0.9 % 2:1 PP 1,000 ml
1,800
Glucose 5 % NaCl 0.9 % 2:1 PP 500 ml
1,800
Glucose 5 % PP 1,000 ml
350
Glucose 5 % PP 250 ml
500
Glucose 5 % PP 500 ml
800
Glucose infusion solution 5 % PP 100 ml
100
Haloperidol injection solution 5 mg i.m./i.v. 5 Amp 1 ml
100
Hydroxyethyl starch 6 % infusion solution 500 ml
2,000
Imipenem/cilastatin 500 mg 10 Amp 20 ml
60
Isoflurane inhalation solution 250 ml
80
iv Line set
15,000
Ketamine injection solution 10 mg/ml 5 vials 20 ml
20
Ketamine injection solution 50 mg/ml 5 vials 10 ml
15
Lactated Ringer’s solution infusion solution 1,000 ml
500
Lactated Ringer’s solution infusion solution 1,000 ml
1,800
Lactated Ringer’s solution infusion solution 500 ml
1,500
Lactated Ringer’s solution infusion solution w/o air 1 L
1,800
Lactated Ringer’s solution infusion solution w/o air 500 ml
600
Lidocaine CO2 injection solution 2 % 10 Amp 20 ml
30
Lidocaine injection solution 1 % 10 Amp 5 ml
400
Lidocaine injection solution 2 % 5 ml w/o cons 10 Amp
120
Lorazepam 20Tbl 1 mg
400
Lorazepam injection solution 4 mg/ml i.v. 10 Amp
75
Mefenamic acid 500 mg 100 Tbl
50
Mepivacaine HCl 10 mg/ml 100 ml
800
Mepivacaine HCl 10 mg/ml 50 ml
500
Mepivacaine HCl 20 mg/ml 50 ml
100
Metamizole sodium injection solution 50 % i.m./i.v. 10 Amp 2 ml
500
Metronidazole infusion solution 500 mg 100 ml
1,400
Midazolam injection solution 15 mg/3 ml i.m./i.v. 5 Amp
80
Midazolam injection solution 5 mg/ml i.m./i.v. 10 Amp
100
Midazolam injection solution 50 mg/10 ml i.m./i.v. 5 Amp
50
Morphine HCl 10 mg/ml 10 Amp 1 ml
500
NaCl 0.9 % irrigation 1,000 ml
600
NaCl 0.9 % irrigation 250 ml
1,400
NaCl 0.9 % infusion solution 1 L
1,800
NaCl 0.9 % infusion solution 100 ml
15,000
NaCl 0.9 % infusion solution 250 ml
300
NaCl 0.9 % infusion solution 500 ml
100
NaCl 0.9 % infusion solution w/o air 1 L
600
NaCl 0.9 % infusion solution w/o air 500 ml
1,000
Norepinephrine/noradrenaline injection solution 0.1 % 10 Amp 10 ml
10
Norepinephrine/noradrenaline injection solution 0.1 % 10 Amp 1 ml
80
Pancuronium bromide injection solution 2 mg/ml 50 Amp 2 ml
50
Pentothal sodium 2.5 g 12 vials
8
Piperacillin/tazobactam 2.25 g vial
120
Propofol injection solution 1 % 5 vials 20 ml
300
Propofol injection solution 1 % vial 50 ml
10
Propofol injection solution. 2 % vial 50 ml
1,500
PVP iodine 10 Gauze pads 7,5 × 22.5 cm
20
PVP iodine alcoholic solution 5 × 1,000 ml
75
PVP iodine solution standardised 500 ml
150
PVP iodine solution standardised120 ml
250
Ringer’s solution irrigation 1,000 ml
1,600
Rocuronium bromide injection solution 50 mg 12 vials
15
Ropivacaine injection solution 0.2 % 5 Bag 200 ml
25
Ropivacaine injection solution 0.75 % 5 Amp 20 ml
25
Sevoflurane liquid 250 ml
60
Silver sulfadiazine cream 50 g
100
Silver sulfadiazine cream 500 g
10
Succinolated Gelatine infusion solution 500 ml
80
Sulfamethoxazole/trimethoprim forte 10 Tbl.
150
Sulfamethoxazole/trimethoprim injection solution i.v. 10 Amp 5 ml
80
Suxamethonium 100 mg 2 Amp 2 ml
80
Tetanus hyper gamma globulin 250 units syringe 1 ml (or corresponding amount of gamma globulin)
15
As a rule, medicines used in bigger amounts in hospitals are commercially supplied by industry, smaller amounts and ad hoc orders by wholesalers, see Chap. 36. Some tasks delegated to the hospital pharmacist may be fulfilled by centralised services for allied partners due to economic or effectiveness reasons. The frame for this duty has to be flexible enough to attain a fast track distribution within the institution and thus a fast dispensing of medicines to the patient. Thus, not only drug supply, but also the medication process and consequently the prevention of medications errors, which are multidisciplinary processes within patient care, are to be considered as integral parts of the mandate [4].
The mandate has not changed throughout decades. It has even become more challenging as new pharmacokinetic and pharmacodynamic knowledge has been emerging and as biopharmaceutic relevant characteristics of highly active ingredients and products can be better anticipated (interactions, drug monitoring, adverse drug events). Therefore, in addition to pure logistics, the hospital pharmacist has to focus more and more on rational and economic drug use and to participate in pharmacotherapy and pharmacovigilance. Mainly in hospitals, medicines use is assessed more and more critical and differentiated. The mandate is even enlarged where suitable, according to special skills of the pharmacist.
The traditional role of the hospital pharmacist still covers production, analysis and assessment of the quality and safety of medicines, which includes the whole supply chain from purchase to pharmacotherapy, and even to disposal of wastes of unused drugs. The complex environment of public health is particularly evident in hospitals. Supply, reconstitution, preparation from raw materials or through adapting products and correct use are a matter of multidisciplinary contributions of many professionals to the benefit of the patient. They require a specialisation as well as life-long learning to remain in a strong and competent position within a care team. Graduate pharmacists have to pass a post-graduate specialisation to get ready to cope with challenges and tasks, which are inherent to the hospital domain (see Sect. 25.4.2).
Fundamental changes and new challenges have been emerging in the last three decades as a result of the globalisation of markets and of production, of new economic doctrines as well as of the development of information technology. This has brought with it a shift in the security of supply and in the hospital pharmacist’s requested and mandatory tasks (Table 3.2).
Table 3.2
Weighting over time of hospital pharmacists’ contributions to shared responsibility and improved outcomes
1960 | 1980 | 2000 | 2020 | |
|---|---|---|---|---|
Clinical pharmacy | + | ++ | +++ | +++ |
Production, quality control, quality assurance | +++ | ++ | ++ | +++ |
Provision and supply chain | ++ | + | ++ | +++ |
Special tasks according to individual skills | + | ++ | +(+) | ++ |
In the past 20 years, investments have often been called off in favour of outsourcing to keep fixed costs small and to optimise balances. From the 1990s, financial interests dominated more and more the patient-centred outcome objectives defended by physicians, pharmacists, other health care professionals and the patient himself. The freedom of action for the hospital pharmacist had been redefined and became more restricted.
Today, the hospital pharmacist is still mandated to control the supply chain, however there are challenges from supply chain professionals supported by those who consider medicines to be no different from any other commodity. Procurement now has to take place in an environment that has more and more budget restraints and external paralysing constraints induced by unwanted dependencies of third party suppliers and sometimes even drug shortages. If the skills are lost, important financial resources would be needed to reactivate lost know-how related to neglected technical equipment or outsourced activities. However, such time is not available in situations such as medicines shortages. Consequently, re-adaption may cost more than ever could have been saved.
3.2.2 Medicines Shortages (Also Referred to as Drug Shortages)
Shortages as a global phenomenon grew steadily and increased sharply in the USA within a few years from 2006 (70 shortages) to 2011 (267 shortages) [5–10]. It is a phenomenon that if left alone threatens to become a crisis in terms of delivering patient care. In 2012, 99 % of over 300 respondents from 27 European countries had to cope with medicines shortage problems according to a survey of the EAHP. Sixty three percent of hospital pharmacists experienced it weekly, sometimes even daily. Seventy seven percent report a worsening of the problem. In Belgium, some 30 medicines are regularly in short supply [11]. Today, in Europe not only isolated cases are in the focus, but examples representing all of the therapeutic groups. In the Netherlands, they are monitored and published on a website. From 2004 to 2011, more than 1,400 products were published. The number increased from 91 in 2004 to 242 in 2011. The duration of a shortage increased from 139 to 242 days in the same period. Substitution (62 %), alternatives (25 %) and pharmacy preparation (2 %) have been the method of choice to cope with such situations [12]. One of the biggest Swiss university hospitals experienced 172 cases of medicines shortage in 2011, i.e. 3 cases per week, with the involvement of 51 suppliers, and with multiple shortages for some products. An out of stock medicines was not available between 21 and 335 days. Classic alkylating, anti-metabolic or topoisomerase-inhibiting antineoplastics with a long time market presence and vaccines are the products for which there is the most concern on the steadily growing list. Pharmaceutical expertise succeeded in finding a suitable solution in 90 % of all cases [13, 14]. To bridge a gap arising from a case of medicines shortage will take 1–7 h [15]. In any case, as a medicine from the hospital formulary has been selected due to a favourable cost – benefit ratio, alternatives are in general cost-intensive compared to the standard product. A simple intermediate substitution of a medicine on the formulary costs 1,800 €, a definite substitution between 3,800 € and 4,690 € (figures from Germany) [16].
Small markets are particularly sensitive to shortages. High registration and regulation affairs cost for market admission may tempt suppliers to economise in countries with low volumes of sales. This is a major problem in a small country. Withdrawal from the market in a country such as Switzerland may be an alert for an upcoming critical situation in the European Union.
In 2011, the situation prompted authorities to intervene in the market and remind manufacturers and suppliers on their responsibility. US President Obama signed the Executive Order 13,588 instructing the FDA to require from manufacturers adequately advanced notices of discontinuation of certain prescription medicines and to review more quickly modifications of the production processes of these medicines [17]. These requirements comprised an obligation to notify and inform on medicines shortages, but do not include a disclosure of the reasons nor of the decisions which lead to a withdrawal of products from the market. An adequate announcement is requested in cases where only one provider for a medically necessary active ingredient is available. The FDA has created a task force for a strategic planning [18] and the EMA reflects particularly on shortages caused by GMP compliance problems [19]. As a result, 38 shortages could be prevented in 2010, 195 in 2011, and 150 in 2012 (up to November), but more has to be done to obtain a sustainable troubleshooting [20].
Relief may arise from less restricted importation frames. Import options depend on the current national legislation and are always related to a lag time for delivery, if substitution cannot be an option. For example, Swissmedic, may temporarily approve imports of EMA-admitted medicines from another European country for an intermediate interval of time in which the local supply chain is interrupted. There are further disadvantages related to importation, in addition to the extra administrative effort. The importing country may be causing a shortage in the exporting country if they are prepared to pay a higher price. In some countries, an imported product can be excluded from reimbursement, if the assurance company is not in agreement.
The most severe among a list of multifactorial reasons [21] which have induced a medicines shortage, were:
Quality or availability problems related to active ingredients or to production processes or equipment (e.g. heparin contamination [22] and propofol case [23])
Demand spikes (e.g. oseltamivir following flu pandemic scenarios [24])
Unintended consequences of contracting by large buyers leading to the loss of small suppliers
Overstocking due to panic buying (especially when alternatives are lacking)
Discontinuation decisions taken by industry, possibly related to pricing or other macro-economic factors (like high cost and low gain)
Globalisation of supply chains creating new vulnerabilities
Lacking alternatives
The latter may be explained by the fact that capital bound in a stock is considered as an important item with potential to optimise a financial balance. The risk of losing capital is reinforced by the availability of new technologies and new products, which might diminish or degrade the stock’s value due to a loss of demand for old products. However, medicines are not comparable to electronic or technical devices with short half-lives. There is no doubt that general economic rules are hardly applicable, one to one, for medicines and in no way for special product groups such as antidotes, narcotics, antineoplastics, total parenteral nutrition, and anti-infectives, if no equivalent and equally expensive medicine is available. Thus, commercial items and lean production are not convincing arguments for small stocks.
It is obvious that most drugs in short supply represent highly active ingredients and the shortage is linked to safety and quality issues. Deviations from GMP uncovered on inspections requiring improvements and investments in a manufacturing plant may play an important role in decision making about maintaining production or not. The risk and the consequences for the supply chain, which arises from cases of a major quality problem and paralysis of a big manufacturing plant after a merger of several smaller sites, is the more threatening as less alternatives will be available. The risk of affecting a global market will be clearly higher in case of one big facility affected instead of many smaller ones. It is even worse, if production is relocated into “low-cost” countries, which have less or no experience in a reliable industrial production free from major operational disruptions. From a delivery, security and ethical point of view, the economic pressure on medicines production has lead to a disastrous situation, which is to everyone’s disadvantage (clinical, financial and health outcomes). An option for pharmacies to immediately cope with the vacuum caused by a stop of industrial manufacturing is only possible if the equipment and quality assurance of its production is regularly updated and the capacity of those still able to produce is sufficient to cover also the needs of non-producing pharmacies.
The role of pharmacists to cope with drug shortages is a determining one if consequences such as decreased safety and worse outcome is to be prevented. The Swiss Association of Public Health Administration and Hospital Pharmacists (GSASA) has edited guidelines to cope with drug shortages [27] and, supported by the most important Swiss Associations and Federations of pharmacists (Swisspharma), physicians (FMH), and hospitals (H+), has signed an agreement with the leading associations of pharmaceutical industry (ASSGP, Intergenerica, Interpharma, Scienceindustries, and Swiss Association of Importers of Proprietary Medicines (VIPS)) to readily provide pharmacies with active ingredients for extemporaneous individualised preparations and small scale stock production of commercially not available formulations or dosages [28]. Whatever the reason for a shortage may be, adaptation from both sides is highly recommended, i.e. from the supplier and from the supply chain responsible in a hospital.
All pharmacies should have an up to date, written policy for managing shortages [29, 30]. That policy should include the need for a risk assessment, which will assess the impact of the shortage and the actions that should be taken to limit those effects. Pharmacists have a responsibility not to do anything that will exacerbate a shortage situation. They have a responsibility to co-operate with any nationally agreed scheme to reduce the effect of such shortages.
3.2.3 Bioequivalence Considerations for Coping with Shortages
Substitution or alternatives, which may be required in the absence or unavailability of appropriate medicinal products on the market, are indispensable to cover the need arising from medicine shortages.1
Generic substitution is defined as the mutual substitution of medicinal products having the same active ingredient, the same strength, and the same dosage form. Different salt forms of the same medicinal product are considered to be the same active substance, unless the salt forms in question exhibit substantial differences in terms of efficacy and activity. Generic substitution usually involves replacing the proprietary brand or reference medicinal product with a generic or parallel-imported product.
The term pharmaceutical alternative is used to define the medicinal product with the same active ingredient, although the dosage form, salt form or strength may vary, such as substitution from a tablet with immediate release to controlled-release, or from capsule to oral solution. Therapeutic substitution is the mutual substitution of medicinal products with different active ingredients, both of which may or may not belong to the same therapeutic group.
In general, medicines, which passed bioequivalence testing, should be substitutable with their generically equivalent, when needed. The European Medicines Agency (EMA) and the Food and Drug Administration (FDA) consider products to be bio-equivalent if, based on the same molar dose, a generic substitute or pharmaceutical alternative exhibits a similar rate and degree of availability at the site of action, and can thus be said to have a similar efficacy and degree of safety.
Market approval of generic medicines requires pharmacokinetic bioequivalence studies. In bio-equivalence studies, the product to be investigated is compared to an innovator product. Products are regarded as bio-equivalent if the 90 % confidence interval of the AUC-ratio and Cmax are within 80–125 % of the reference product. If the confidence interval is within these limits, this means that the average will deviate far less from the corresponding value found for the innovator product.
For medicinal products with a narrow therapeutic index the 90 % confidence interval of the AUC ratio must lie between 90.00 % and 111.11 %, and if Cmax is important then this too must lie between 90.00 % and 111.11 %. The significance of this, in terms of interchangeability, is not known.
For medicinal products with large intra-individual variation (i.e. if the variation of a kinetic parameter exceeds 30 %) the 90 % confidence interval of Cmax should be between 69.84 % and 143.19 %, while the AUC-ratio should be within normal limits. Classical bioequivalence studies have limited value in indicating equivalent efficacy and safety for biosimilars (generic version of biological medicines).
Medicines with a different dosage form are not tested for bioequivalence. These medicines have different kinetic properties and they are not bioequivalent by itself. From that viewpoint, these products cannot be substituted and caution is needed. The consequences for non-adherence and non-efficacy should be considered.
The main consideration where generic substitution is concerned with is that the efficacy and safety of substituted medicinal products should be equivalent to one another. As this is tested during the approval of generic medicinal products, on the basis of bioequivalence studies, it can be assumed that the approved generic products are just as effective and safe as the reference product. However, in conjunction with certain active substances or certain situations, it may be preferable to avoid even the slightest risk (e.g. ciclosporine).
In addition, there is a range of other issues – unrelated to bioequivalence – which can cause problems following substitution. Accordingly, there is still a need to determine the advisability of substitution on a medicine-by-medicine and patient-by-patient basis. The flow chart in Fig. 3.1 and the following directions may be helpful in this regard:
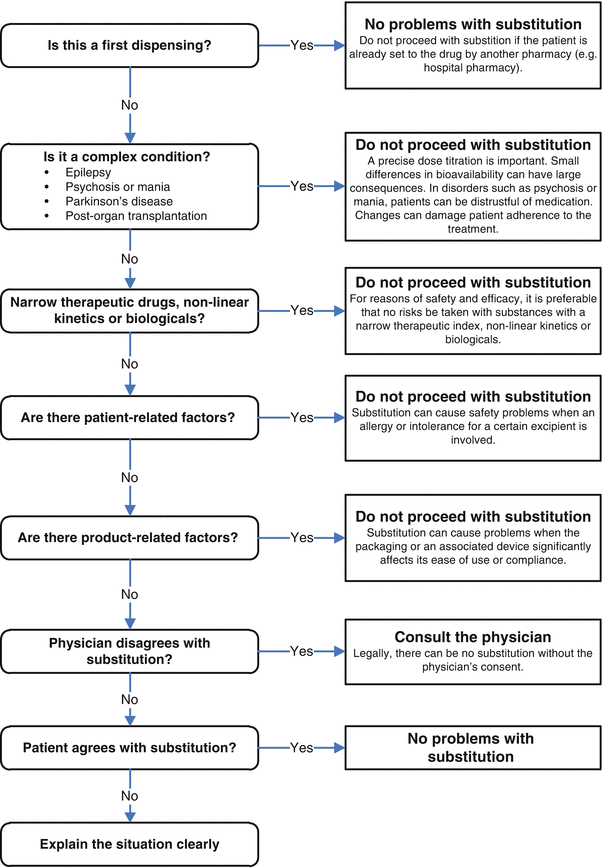
Fig. 3.1
Flowchart with decision points for substitution. Adapted from [31] by the same author. For substitution bioequivalence and additional factors are considered, especially if repeat dispensing is required. Bioequivalent proven medicinal products should be substitutable. However, for a variety of reasons this may not be the case and caution needs to be warranted
(a)
On first dispensing, the problems relating to efficacy, safety and convenience for the patient are not an issue. This is not the case if the patient has already received the medicine from another pharmacy (e.g. hospital pharmacy).
(b)
A precise dose titration is important. Small differences in bioavailability can have large consequences. In disorders such as psychosis or mania, patients can be distrustful of medication. Changes can damage patient adherence to the treatment.
(c)
Substances with which, for reasons of safety and efficacy, it is preferable that no risks be taken are biologicals, those with a narrow therapeutic index and those with non-linear kinetics. Although substances with a narrow therapeutic index or non-linear kinetics meet the requirements for bioequivalence, and are therefore theoretically interchangeable, patient-related factors that adversely affect interchangeability may be involved.
(d)
Substitution can cause safety problems when the medicinal products contains an excipient to which the patient is allergic or intolerant.
(e)
The packaging of the medicinal product in question, or an associated device, significantly affects its ease of use, or compliance.
(f)
Legally, there can be no substitution without the physician’s consent, unless prescribed generically. In practice, this usually means that agreements have been made on this point.
3.3 Medicines with a Market Authorisation
3.3.1 Market Authorisation (Formerly “Registration”)
In Europe as in many other parts of the world, medicines can only be marketed if they are authorised [32–34]. A company, which wants to market a medicinal product, has to apply for a marketing authorisation at the European Medicines Agency for the European Union [35] or at the Medicines Agency of a country. The Medicines Agencies scientifically evaluate the medicine and grant an application if they have safety, quality and efficacy assessed positively. The process, which formerly was called registration of medicines, now is to be spoken of as granting of a Marketing Authorisation. And the company is the Marketing Authorisation Holder (MAH).
The applicant has to be authorised for manufacture, import, wholesaling, export or trading in foreign countries, according to the activities and the locations of the business. The applicant has to submit a product dossier with all necessary data defined in a guideline [36]. Such an authorisation is limited in time. It is renewed after an inspection. It nominates the Qualified Persons and specifies limitations or conditions. To be allowed to produce a medicinal product the manufacturer or the importer needs a Manufacturing License, which is bound to compliance to GMP (see Sect. 35.5.2). If the Medicines Agency judges positively, the European Commission or the National Authorities grant a Marketing Authorisation for the entire European economic area (EEA; EU Member States plus Switzerland, Norway, Iceland and Liechtenstein) or just for the country itself. Conversely, local authority’s approval does not grant any authorisation for other EU member states. Non Member States ratify EU legislation such as on the pharmacopoeia to adapt national legislation and may have treaties with the EU, USA, Australia or Singapore [37, 38] European registration is possible for all medicines which meet certain requirements [39]. It is however compulsory for specific medicinal products such as biotechnologicals, orphan medicinal products, anti-neoplastics or medicines for autoimmune diseases. The product is recognisable by a EU-authorised medicinal product registration number (for example: EU/1/04/276/001).
Product information on European authorised medicines can be found at the EMA Regulatory and procedural guidance index [40]. This information comprises:
A list of authorised presentations of the medicinal product
The summary of product characteristics (SmPC)
The patient leaflet and labelling of the product
The European Public Assessment Report (EPAR)
A medicinal product with a national marketing authorisation has a national registration number, e.g. RVG 11,985 in the Netherlands. Product information about nationally authorised medicines can normally be found on national websites. National Medicinal Agencies refer to the website of the EMA if the product has obtained a European Marketing Authorisation.
3.3.2 Reimbursement
The manufacturer is allowed to market a product with a Marketing Authorisation. The company sets the price of the medicine. This is done either by a calculation which takes into account the manufacturing and marketing costs, including a profit allowance, or it is set in comparison to competing products of the same kind, especially, if the authorities negotiate with the company about that price. National pricing and financing policies are guided by a WHO policy [41].
Regulations for reimbursement are still nationally determined. In many countries the approaches are more or less the same: the type of health insurance system, pharmacoeconomic data, the effectiveness of the medicine, and the need in relation to similar medicines are determinant. The cost of a medicine for hospital patients may be regulated differently from the community situation. The inclusion in clinical guidelines of a specific medicine is of major importance in order to obtain reimbursement.
The key questions by the assessor are about an added benefit and about the medical value. In the Netherlands, the medical value is assessed unofficially by means of the Dunnings Funnel, which evaluates the candidates by defined criteria, e.g. necessity, effectiveness, safety, cost-effectiveness commonly calculated as incremental cost-effectiveness ratio (ICER), and social arguments such as budget impact or own responsibility [42]. The societies’ willingness to pay for an additional quality-adjusted life year gained (QALY) is as follows [43–45]:
Canada: $ 20,000 – $ 100,000
United States: $ 50,000 – $ 100,000
The Netherlands: € 20,000 – € 50,000
Belgium: € 50,000
United Kingdom: £ 20,000 – £ 30,000
WHO standard: 3 * GDP (gross domestic product) per capita
The added medical benefit may be assessed in comparison with existing therapies in terms of effectiveness, adverse effects, experience, applicability and ease of use. In France, the first step of reimbursement decision and price fixing process is confirming the medical benefit obtained (SMR, service medical rendu) which determines the reimbursement percentage, whereas the second step evaluates the improvement of the medical benefit over existing medicines (ASMR, amélioration du service médical rendu), which is used for price negotiations [46]. In contrast to the methods of healthcare evaluation in other countries, the UK National Institute for Health and Care Excellence (NICE) does not evaluate all interventions as they reach the market. NICE has published guidelines on how it will select interventions for review. This includes the following key questions [47–49]:
Is the technology likely to result in a significant health benefit, taken across the National Health Service (NHS) as a whole, if given to all patients for whom it is indicated?
Is the technology likely to result in a significant impact on other health related government policies (e.g. reduction in health inequalities)?
Is the technology likely to have a significant impact on NHS resources (financial or other) if given to all patients for whom it is indicated?
Is the institute likely to be able to add value by issuing national guidance?
Many countries, e.g. Switzerland, have compulsory social accident and health insurance systems for every citizen. The choice of the insurance company is free. The insurer has to accept every request and is not allowed to reject applicants with increased risks in the basic part. Rejection is only possible for coverage by complementary insurances. Physicians, pharmacists, midwives, chiropractors, laboratories, hospitals, several institutions for acute or chronic care for in- or outpatients, policlinics, or ambulance transporters are care providers approved from the concordat of insurers. Care providers are licensed to bill the insurer for approved services at prefixed rates according to lists such as TARMED and SwissDRG (German modification) issued by the Federal Office of Public Health (FOPH) [50, 51]. To be put on the list of pharmaceutical specialties, a request has to be addressed to the Swiss Federal Social Insurance Office, which is advised by the Swiss Federal Drug Commission. Applicants have to follow a manual and submit several documents, e.g. a summary of product characteristics, the grant of marketing authorisation, key facts, clinical overview, non-clinical overview, most relevant clinical studies, epidemiologic data of the disease to be treated, clinical guidelines, and pharmacoeconomic studies [52]. It is stipulated in the Swiss federal act on health insurances, that medicines and care are required to be efficacious, appropriate and economic to be reimbursed [53]. The latter requirement is checked by means of price comparisons between the requested Swiss price and those applied in Denmark, Germany, the Netherland, Great Britain, France, and Austria [52].
The costs of materials, duration of preparation, quality control, investment in premises, training, quality assurance et cetera determine the basic cost of pharmacy preparation. As with the reimbursement of licensed medicines there is a distinction between in-patient and out-patient supply. Pharmacy preparations used in hospitals could be considered to be part of the reimbursement for the therapy as a whole. Anyhow, the hospital pharmacist normally has to find his payment within the hospital organisation. In community pharmacy most pharmacy preparations are reimbursed by the health insurer, according to the Tax price with a surcharge according to the performance cost system. Reimbursement for unlicensed medicines will be handled differently in most European Countries. There may also be differences between the reimbursement for hospital and public pharmacies.
Within the patient access and reimbursement schemes, risk sharing is fixed as outcome-based or financial-based agreement between the payer and the manufacturer. Financial-based agreements are possible on a fixed price, on a price-volume ratio, on a price by diagnosis, on capitation fee, or on dose-quantity limits. Outcome-based agreements can be divided into evidence-development-based, conditional treatment continuation-based, or performance-based schemes. Most of these schemes are applied in Europe and Australia, followed by Canada and the United States [54, 55]:
Evidence development schemes (34 schemes in use, regrouped into coverage-with-study or coverage-with-appropriateness determination approaches)
Example taxanes: In 2000 in the UK, the use of taxanes for adjuvant treatment of early breast cancer was limited to randomised clinical trials.
Example temozolomide: In 2001 in the UK, this active ingredient was only recommended as an initial chemotherapy for patients with brain cancer included in a clinical trial.
Example risperidone: In 2003 in France, costs were covered, if evaluation studies on whether it helps patients stay on the medications were performed. In case of failure the manufacturer was to refund costs to the French ministry of health.
Example human papilloma virus quadrivalent vaccine: In 2007 in Sweden, the manufacturer was asked to provide every 6 months additional data on ongoing and planned studies in order to determinate the cost-effectiveness from a long-term perspective.
Conditional treatment continuation schemes (10 schemes in use)
Example bortezomib: For the multiple myeloma indication, in the UK the manufacturer agreed in 2007 to reimburse the NHS in either cash or product for patients who did not respond, i.e. those who do not show a 50 % decrease in serum M protein, after four cycles. Responding patients received additional four cycles. In 2009, the same agreement was fixed with the Scottish Medicines Consortium.
Examples sunitinib and sorafenib: A hospital discount of 50 % applies in Italy to the first 3 months of treatment. For responding patients the treatment is then reimbursed and the discount dropped.
Examples Alzheimer’s disease medicines: In Italy, during the first 3 months, patients starting Alzheimer’s disease medicines are assessed for short-term effectiveness. The medicines are provided free of charge by the manufacturer. If treatment goals are met after 3 months, treatment is continued for a maximum of 2 years and the costs reimbursed by the Italian Drugs Agency (AIFA).
Performance-linked reimbursement schemes (14 schemes in use, regrouped into pricing review, try-before-you-buy, or no cure – no pay principles)
Example statins: In 1998 in the US and in 2000 in the UK, rebates were agreed and refunds were promised if LDL cholesterol could not be lowered.
Example bosentan: In 2004 in Australia, the price of bosentan for pulmonary arterial hypertension was linked to the survival of patients followed in an observational study.
Example risedronate sodium: In the US in 2009, the manufacturer agreed to reimburse for the costs of treating-related fractures.
3.4 Investigational Medicinal Products
The manufacturing of investigational medicines goes together with phases I – III of the Clinical Trial Investigation, where pharmacokinetics and toxicology at different dosages is investigated and compared with the standard treatment or placebo treatment in a small group of healthy volunteers first, in a limited group of patients afterwards, and finally in a large group of patients. After completing these investigations the new medicine can be offered for approval and admission to the market (market authorisation). In phase IV Authorised Medicines are evaluated for the authorised indications, side effects and long term value and will be monitored in clinical practice. This may occur by pharmacovigilance or by outcomes research in specific patient populations. As described in Table 3.3, currently 15 years may pass until a new chemical entity reaches the market.
Table 3.3
Phases of clinical research
Discovery | Clinical trials | Launching | ||||
|---|---|---|---|---|---|---|
Preclinical testing | Phase I | Phase II | Phase III | Drug agency | Phase IV | |
Years approximately | 6.5 | 1.5 | 2 | 3.5 | 1.5 | |
Test population | Laboratory and animal studies | 20–100 healthy volunteers | 100–500 patient volunteers | 1,000–5,000 patient volunteers | Review process, approval | Additional post-marketing testing |
Purpose | Assess safety, biological activity and formulations | Determine safety and dosage | Evaluate effectiveness, look for side effects | Confirm effectiveness, monitor adverse reactions from long-term use | ||
Yield | 5,000 compounds evaluated | 5 enter trials | 1 approved | |||
In the clinical phase of development of new chemical entities, medicines are developed by hospitals, universities, or pharmaceutical companies and administered to humans as “Investigational Medicinal Products (IMP)”. In Europe, the administration of IMPs to human beings is regulated by Directive 2001/20/EC (which has been replaced on the 16 April 2014 by the new Regulation No 536/2014 which is to come into force no earlier than 28th May 2016), which deals with the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use [56]. Each specific investigation has to be approved by an Ethics Committee. In the Netherlands, a national committee for clinical research has to assign a certificate of incorporation. Research with a non-licensed medicine without such an approval is not allowed. In Switzerland, a new act on human research entered into force as from 2014. It inserts an article into the Swiss Federal Constitution, recently voted and approved by the Swiss nation in 2010 [57].
The dossier of an IMP is called the Investigational Medical Product Dossier (IMPD). It describes the technological, pharmacological and toxicological properties of the product as well as the method of preparation. Importing or preparing IMPs by a manufacturer or a pharmacist requires a license/authorisation [58]. An authorisation as a wholesale trader in medicinal products is required, if the IMP originates from another ERA state (European Research Area). A Manufacturing Authorisation is requested, if the medicine is imported from a country outside the ERA. These authorisations are specific for a dosage form or for a preparation process. Preparation and quality control should be performed according to the IMPD. A Qualified Person (QP, see Sect. 25.3.4) has to release the product after import, preparation and quality control and to guarantee that all quality requirements are met. Pharmacies don’t need a Manufacturing License if the preparation of an IMP is limited to operations such as reconstitution, dilution and labelling, which have to be performed for the purpose of administration to the patient and are defined in the IMPD. These activities however have to be carried out within the institution where this clinical trial is carried out and by a pharmacist who is employed within this institution [59]. See also Sect. 35.5.10.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


