Assessment of biopharmaceutical properties
Marianne Ashford
Chapter contents
Measurement of key biopharmaceutical properties
Release of drug from its dosage form into solution
Stability in physiological fluids
Plasma concentration-time curves
Cumulative urinary drug excretion curves
Absolute and relative bioavailability
Assessment of site of release in vivo
Biopharmaceutics Classification System
Key points
Introduction
Biopharmaceutics is concerned with factors that influence the rate and extent of drug absorption. As discussed in Chapters 19 and 20, the factors that affect the release of a drug from its dosage form, its dissolution into physiological fluids, its stability within those fluids, its permeability across the relevant biological membranes and its presystemic metabolism will all influence its rate and extent of absorption (Fig. 21.1). Once the drug is absorbed into the systemic circulation, its distribution within the body tissues (including to its site of action), its metabolism and elimination are described by the pharmacokinetics of the compound (discussed in Chapter 18). This in turn influences the length and magnitude of the therapeutic effect or the response of the compound, i.e. its pharmacodynamics.
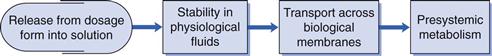
The key biopharmaceutical properties that can be quantified and therefore give an insight into the absorption of a drug are its:
• release from its dosage form into solution at the absorption site
• stability in physiological fluids
As most drugs are delivered via the mouth, these properties will be discussed with respect to the peroral route. The bioavailability of a compound is an overall measure of its availability in the systemic circulation and so the assessment of bioavailability will also be discussed. Other methods of assessing the performance of dosage forms in vivo will also be briefly mentioned. The Biopharmaceutics Classification System (BCS), which classifies drugs according to dose and two of their key biopharmaceutical properties, solubility and permeability, is outlined.
Measurement of key biopharmaceutical properties
Release of drug from its dosage form into solution
As discussed in Chapter 20 and Part 5 of this book, a dosage form is normally formulated to aid and/or control the release of drug from it. For example, for an immediate-release tablet, the tablet needs to disintegrate to yield the primary drug particles. Further, a suspension should not be so viscous that it impedes the diffusion of dissolving drug away from the solid particles.
The solubility of a drug across the gastrointestinal pH range will be one of the first indicators as to whether dissolution is liable to be rate limiting in the absorption process. Knowledge of the solubility across the gastrointestinal pH range can be determined by measuring the equilibrium solubility in suitable buffers or by using an acid or a base titration method.
Methods of measuring the dissolution rate of both a drug itself (intrinsic dissolution rate) and of various dosage forms are discussed in Chapters 2 and 35, and in the relevant chapters of Part 5.
The aim of dissolution testing is to find an in vitro characteristic of a potential formulation that reflects its in vivo performance. When designing a dissolution test to assess drug release from a biopharmaceutical perspective, it is important to mimic as closely as possible the conditions of the gastrointestinal tract. Clinical scientists increasingly want to rely on dissolution tests to establish in vitro/in vivo correlations between the release of drug from the dosage form and its absorption. If this can be successfully achieved, it is possible that the dissolution test could replace some of the in vivo studies that need to be performed during product development and registration. Such correlations should have the benefits of reducing the use of animals to evaluate formulations and the size and number of costly clinical studies to assess bioavailability as well as being used to allow formulation, process and site of manufacture changes.
An in-vitro/in-vivo correlation may only be possible for those drugs where dissolution is the rate-limiting step in the absorption process. Determining full dissolution profiles of such drugs in a number of different physiologically representative media will aid the understanding of the factors affecting the rate and extent of dissolution. The profiles can also be used to generate an in vitro/in vivo correlation. To achieve this, at least three batches that differ in their in vivo as well as their in vitro behaviour should be available. The differences in the in vivo profiles need to be mirrored by the formulations in vitro. Normally, the in vitro test conditions can be modified to correspond with the in vivo data to achieve a correlation. Very often, a well-designed in vitro dissolution test is found to be more sensitive and discriminating than an in vivo test. From a quality assurance perspective, a more discriminating dissolution method is preferred because the test will indicate possible changes in the product before the in vivo performance is affected.
In-vitro dissolution testing of solid dosage forms is discussed fully in Chapter 35. The reader is referred there for consideration of the apparatus available and suitable dissolution media to simulate as closely as possible gastric and intestinal fluids. This application of dissolution testing is discussed further here in the context of the assessment of biopharmaceutical properties.
A dilute hydrochloric acid-based solution at pH 1.2 can simulate gastric fluid quite closely (but obviously not exactly) and phosphate-buffered solution at pH 6.8 can mimic intestinal fluid. However, dissolution media more closely representing physiological conditions may well provide more relevant conditions. A range of dissolution media that are widely accepted to mimic physiological parameters in gastric and intestinal fluids in the fed and fasted states are available. Each of these media takes into account not only the pH of the fluids in the different states but their ionic composition, surface tension, buffer capacity and bile and lecithin contents. Details of simulated gastric and intestinal fluids for both the fed and fasted state are given in Tables 35.2 and 35.3.
The conditions within the stomach in the fed state are highly dependent on the composition of the meal eaten and are therefore difficult to simulate. In trying to produce an in vitro/in vivo correlation, it has been suggested that a more appropriate way of simulating the fed-state gastric fluids is to homogenize the meal to be used in clinical studies and then dilute it with water. Long-life milk has also been used to simulate gastric conditions in the fed state.
It has been proposed that the duration of the dissolution test should depend on the site of absorption of the drug and its timing of administration. Thus, in designing a dissolution test, some knowledge or prediction of the permeability properties of the drug is beneficial. If, for example, the drug is absorbed from the upper intestine and is likely to be dosed in the fasted state, the most appropriate dissolution conditions may be a short test (~5–30 minutes) in a medium simulating gastric fluid in the fasted state. Alternatively, if it is advised that a drug should be administered with food and the drug is known to be well absorbed throughout the length of the gastrointestinal tract, a far longer dissolution test may be more appropriate. This could perhaps be several hours in duration with a range of media such as, initially, simulated gastric fluid to mimic the fed state, followed by simulated intestinal fluid to mimic both fed and fasted states.
The volumes of fluid within, and the degree of agitation of, the stomach and intestines vary enormously, particularly between the fed and the fasted states. Consequently, it is difficult to choose a representative volume and degree of agitation for an in vitro test. Guidance given to industry on the dissolution testing of immediate-release solid oral dosage forms suggests volumes of 500, 900 or 1000 mL and gentle agitation conditions. Regulatory authorities will expect justification of a dissolution test to ensure that it will discriminate between a good and a poor formulation, and thus see it as a critical quality test in submissions of applications for Marketing Authorizations.
Stability in physiological fluids
The stability of drugs in physiological fluids (in the case of orally administered drugs, the gastrointestinal fluids) depends on two factors:
Means of assessing the chemical stability of a drug are discussed in Chapters 48 and 49. The stability of a drug in gastrointestinal fluids can be assessed by simulated gastric and intestinal media or by obtaining gastrointestinal fluids from humans or animals. The latter provides a harsher assessment of gastrointestinal stability but is more akin to the in vivo setting. In general, the drug is incubated with either real or simulated fluid at 37 °C for a period of 3 hours and the drug content analysed. A loss of more than 5% of drug indicates potential instability. Many of the permeability methods described below can be used to identify whether gastrointestinal stability is an issue for a particular drug.
For drugs that will still be in the gastrointestinal lumen when they reach the colonic region, resistance to the bacterial enzymes present in this part of the intestine needs to be considered. The bacterial enzymes are capable of a whole host of reactions. There may be a significant portion of a poorly soluble drug still in the gastrointestinal tract by the time it reaches the colon. If the drug is absorbed along the length of the gastrointestinal tract, and is susceptible to degradation or metabolism by the bacterial enzymes within the tract, the drug’s absorption and hence its bioavailability is liable to be reduced. Similarly, for sustained- or controlled-release products that are designed to release their drug along the length of the gastrointestinal tract, the potential of degradation or metabolism by bacterial enzymes should be assessed. If a drug is metabolized to a metabolite which can be absorbed, the potential toxicity of this metabolite should be considered.
Permeability
There is a wealth of techniques available for either estimating or measuring the rate of permeation across membranes that are used to gain an assessment of oral absorption in humans. These range from computational (in silico) predictions to both physicochemical and biological methods. The biological methods can be further subdivided into in vitro, in situ and in vivo methods. In general, the more complex the technique, the more information that can be gained and the more accurate is the assessment of oral absorption in humans. The range of techniques is summarized in Table 21.1. Some of the more widely used ones are discussed below.
Table 21.1
Some of the models available for predicting or measuring drug absorption
| Model type | Model | Description |
| Computational/In silico | clog P | Commercial software that calculates n-octanol/ water partition coefficient based on fragment analysis, known as the Leo-Hansch method |
| mlog P | Method of calculating log P, known as the Moriguchi method (see text) | |
| Physicochemical | Partition coefficient | Measure of lipophilicity of drug, usually measured between n-octanol and aqueous buffer via a shake-flask method |
| Immobilized artificial membrane | Measures partition into more sophisticated lipidic phase on an HPLC column | |
| Cell culture | Caco-2 monolayer | Measures transport across monolayers of differentiated human colon adenocarcinoma cells |
| HT-29 | Measures transport across polarized cell monolayer with mucin-producing cells | |
| Excised tissues | Cells | Measures uptake into cell suspensions, e.g. erythrocytes |
| Freshly isolated cells | Measures uptake into enterocytes; however; the cells are difficult to prepare and are short-lived | |
| Membrane vesicles | Measures uptake into brush border membrane vesicles prepared from intestinal scrapings or isolated enterocytes | |
| Everted sacs | Measures uptake into intestinal segments/sacs | |
| Everted intestinal rings | Studies the kinetics of uptake into the intestinal mucosa | |
| Isolated sheets | Measures the transport across sheets of intestine | |
| In situ studies | In situ perfusion | Measures drug disappearance from either closed or open loop perfusate of segments of intestine of anaesthetized animals |
| Vascularly perfused intestine | Measures drug disappearance from perfusate and its appearance in blood | |
| In vivo studies | Intestinal loop | Measures drug disappearance from perfusate of loop of intestine in awake animal |
| Human data | Loc-I-Gut | Measures drug disappearance from perfusate of human intestine |
| High-frequency capsule | Non-invasive method; measures drug in systemic circulation | |
| InteliSite capsule | Non-invasive method; measures drug in systemic circulation | |
| Bioavailability | Deconvolution of pharmacokinetic data |
Partition coefficients
One of the first properties of a molecule that should be predicted or measured is its partition coefficient between oil and a water phase (log P). This gives a measure of the lipophilicity of a molecule, which can be used to predict how well it will be able to cross a biological membrane. It is a very useful parameter for many reasons relating to formulation design and drug absorption and is discussed elsewhere in Chapters 2, 20 and 23. As discussed in Chapter 20, n-octanol is most commonly chosen as the solvent for the oil phase as it has similar properties to biological membranes although other oil phases have been used (as considered in Chapter 23). One of the most common ways of measuring partition coefficients is to use the shake flask method (Fig. 21.2). It relies on the equilibrium distribution of a drug between oil and an aqueous phase. Prior to the experiment the aqueous phase should be saturated with the oil phase and vice versa. The experiment should be carried out at constant temperature. The drug should be added to the aqueous phase and the oil phase which, in the case of n-octanol, as it is less dense than water, will sit on top of the water. The system is mixed and then left to reach equilibrium (usually at least 24 hours). The two phases are separated and the concentration of drug is measured in each phase and a partition coefficient calculated. This technique is discussed further in the context of preformulation in Chapter 23.
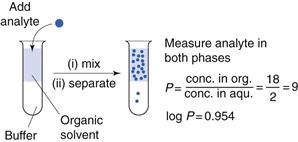
If the aqueous phase is at a particular pH, the distribution coefficient at that pH is measured (log D); this then accounts for the ionization of the molecule at that pH. In the case of a weakly acidic or a weakly basic drug, the log D measured at an intestinal pH (e.g. 6.8) is liable to give a better prediction of the drug’s ability to cross the lipid gastrointestinal membrane than its partition coefficient, log P, which does not take the degree of ionization into account.
As discussed in Chapter 20, within a homologous series, increasing lipophilicity (log P or log D) tends to result in greater absorption. A molecule is unlikely to cross a membrane (i.e. be absorbed via the transcellular passive route) if it has a log P less than 0.
Instead of determining log P experimentally, computational methods can be used to estimate it. There are a number of software packages available to do this. There is a reasonably good correlation between calculated and measured values. Log P can be estimated by breaking down the molecule into fragments and calculating the contribution of each fragment to overall lipophilicity (often called the clog P). Another way of estimating log P is the Moriguchi method, which uses 13 parameters for hydrophobic and hydrophilic atoms, proximity effects, unsaturated bonds, intramolecular bonds, ring structures, amphoteric properties and several specific functionalities to obtain a value for the partition coefficient. This is often called the mlog P. The advantages of these methods are in drug discovery, where an estimate of the lipophilicity of many molecules can be obtained before they are actually synthesized.
Another, more sophisticated physicochemical means of estimating how well a drug will partition into a lipophilic phase is by investigating how well the molecule can be retained on a high-performance liquid chromatography (HPLC) column. HPLC columns can be simply coated with n-octanol to mimic n-octanol-aqueous partition or, more elaborately, designed to mimic biological membranes. For example the immobilized artificial membrane (IAM) technique provides a measure of how well a solute (i.e. the drug) in the aqueous phase will partition into biological membranes (i.e. be retained on the column). Good correlations between these methods and biological in vitro methods of estimating transcellular passive drug absorption have been obtained.
Cell culture techniques
Cell culture techniques for measuring the intestinal absorption of molecules have been increasingly used over recent decades and are now a well-accepted model for absorption. The cell line that is most widely used is Caco-2.
Caco-2 cells are a human colon carcinoma cell line that was first-proposed and characterized as a model for oral drug absorption by Hidalgo. In culture, Caco-2 cells spontaneously differentiate to form a monolayer of polarized enterocytes. These enterocytes resemble those in the small intestine, in that they possess microvilli and many of the transporter systems present in the small intestine, for example those for sugars, amino acids, peptides and the P-glycoprotein efflux transporter. Adjacent Caco-2 cells adhere through tight junctions. However, the tightness of these junctions is more like those of the colon than those of the leakier small intestine.
There are many variations on growing and carrying out transport experiments with Caco-2 monolayers. In general, the cells are grown on porous supports, usually for a period of 15–21 days in typical cell culture medium, Dulbecco’s Modified Eagle Medium supplemented with 20% foetal bovine serum, 1% non-essential amino acids and 2 mM L-glutamine. The cells are grown at 37 °C in 10% carbon dioxide at a relative humidity of 95%. The culture medium is replaced at least twice each week. Transport experiments are carried out by replacing the culture medium with buffers, usually Hank’s Balanced Salt Solution adjusted to pH 6.5 on the apical surface and Hank’s Balanced Salt Solution adjusted to pH 7.4 on the basolateral surface (Fig. 21.3).
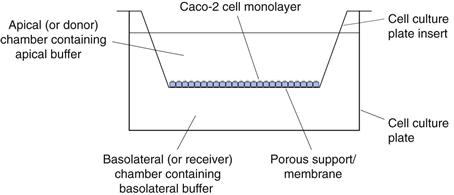
After a short incubation period, usually about 30 minutes, when the cells are maintained at 37°C in a shaking water bath, the buffers are replaced with fresh buffers and a dilute solution of drug is introduced to the apical chamber. At regular intervals, the concentration of the drug in the basolateral chamber is determined. The apparent permeability coefficient across cells can be calculated as follows:
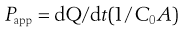 (21.1)
(21.1)
where Papp is the apparent permeability coefficient (cm/s), dQ/dt is the rate of drug transport (µg s−1), C0 is the initial donor concentration (mg/mL) and A is the surface area of the monolayer (cm2).
To check that the monolayer has maintained its integrity throughout the transport process, a marker for paracellular absorption, such as mannitol, which is often radiolabelled for ease of assay, is added to the apical surface. If less than 2% of this crosses the monolayer in an hour then the integrity of the monolayer has been maintained. Another way to check the integrity of the mono-layer is by measuring the transepithelial resistance (TER).
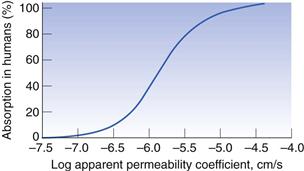
To use the Caco-2 cells as an absorption model, a calibration curve needs to be generated. This is done for compounds for which the absorption in humans is known. Figure 21.4 shows the general shape of the curve of fraction absorbed in humans versus the apparent permeability coefficient in Caco-2 cells. As cells are biological systems, small changes in their source, method of culture and the way in which the transport experiment is performed will affect the apparent permeability of a drug, such that this curve can shift significantly to the right or left, or alter in its gradient. Therefore, when carrying out Caco-2 experiments, it is important always to standardize the procedure within a particular laboratory and ensure that this procedure is regularly calibrated with a set of standard compounds.
Caco-2 monolayers can also be used to elucidate the mechanism of permeability. If the apparent permeability coefficient is found to increase linearly with increasing concentration of drug (i.e. the transport is not saturated), is the same whether the drug transport is measured from the apical to basolateral or the basolateral to apical direction, and is independent of pH, it can be concluded that the transport is a passive and not an active process. If the transport in the basolateral to apical direction is significantly greater than that in the apical to basolateral direction then it is likely that the drug is actively effluxed from the cells by a counter-membrane transporter, such as P-glycoprotein. If the transport of the drug is also inhibited by the presence of compounds that are known inhibitors of P-glycoprotein this gives a further indication that the drug is susceptible to P-glycoprotein efflux.
To help elucidate whether other membrane transporters are involved in the absorption of a particular drug, further competitive inhibition studies can be carried out with known inhibitors of the particular transporter. For example, the dipeptide glycosyl-sarcosine can be used to probe whether the dipeptide transporter is involved in the absorption of a particular drug.
To evaluate whether a compound is absorbed via the paracellular or the transcellular pathway, the tight junctions can be artificially opened with compounds such as EDTA, which chelates calcium. Calcium is involved in keeping the junctions together. If the apparent permeability of a compound is not affected by the opening of these junctions, which can be assessed by using a paracellular marker such as mannitol, one can assume the drug transport is via a transcellular pathway.
If the disappearance of drug on the apical side of the membrane is not mirrored by its appearance on the basolateral side, and/or the mass balance at the end of the transport experiment does not account for 100% of the drug, there may be a problem with binding to the membrane porous support. This will need investigation, or the drug may have a stability issue. The drug could be susceptible to enzymes secreted by the cells and/or to degradation by hydrolytic enzymes as it passes through the cells, or it may be susceptible to metabolism by cytochrome P450 within the cell. Thus the Caco-2 cells are not only capable of evaluating the permeability of drugs but also have value in investigating whether two of the other potential barriers to absorption, namely stability and presystemic metabolism, are likely to affect the overall rate and extent of absorption.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


