PART 8: Infectious Diseases
SECTION 1 | BASIC CONSIDERATIONS IN INFECTIOUS DISEASES |
144 | Approach to the Patient with an Infectious Disease |
HISTORICAL PERSPECTIVE
The origins of the field of infectious diseases are humble. The notion that communicable diseases were due to a miasma (“bad air”) can be traced back to at least the mid-sixteenth century. Not until the work of Louis Pasteur and Robert Koch in the late nineteenth century was there credible evidence supporting the germ theory of disease—i.e., that microorganisms are the direct cause of infections. In contrast to this relatively slow start, the twentieth century saw remarkable advances in the field of infectious diseases, and the etiologic agents of numerous infectious diseases were soon identified. Furthermore, the discovery of antibiotics and the advent of vaccines against some of the most deadly and debilitating infections greatly altered the landscape of human health. Indeed, the twentieth century saw the elimination of smallpox, one of the great scourges in the history of humanity. These remarkable successes prompted noted scholar Aidan Cockburn to write in a 1963 publication entitled The Evolution and Eradication of Infectious Diseases: “It seems reasonable to anticipate that within some measurable time … all the major infections will have disappeared.” Professor Cockburn was not alone in this view. Robert Petersdorf, a renowned infectious disease expert and former editor of this textbook, wrote in 1978 that “even with my great personal loyalties to infectious diseases, I cannot conceive a need for 309 more [graduating trainees in infectious diseases] unless they spend their time culturing each other.” Given the enormous growth of interest in the microbiome in the past 5 years, Dr. Petersdorf’s statement might have been ironically clairvoyant, although he could have had no idea what was in store for humanity, with an onslaught of new, emerging, and re-emerging infectious diseases.
Clearly, even with all the advances of the twentieth century, infectious diseases continue to represent a formidable challenge for patients and physicians alike. Furthermore, during the latter half of the century, several chronic diseases were demonstrated to be directly or indirectly caused by infectious microbes; perhaps the most notable examples are the associations of Helicobacter pylori with peptic ulcer disease and gastric carcinoma, human papillomavirus with cervical cancer, and hepatitis B and C viruses with liver cancer. In fact, ~16% of all malignancies are now known to be associated with an infectious cause. In addition, numerous emerging and re-emerging infectious diseases continue to have a dire impact on global health: HIV/AIDS, pandemic influenza, and severe acute respiratory syndrome (SARS) are but a few examples. The fear of weaponizing pathogens for bioterrorism is ever present and poses a potentially enormous threat to public health. Moreover, escalating antimicrobial resistance in clinically relevant microbes (e.g., Mycobacterium tuberculosis, Staphylococcus aureus, Streptococcus pneumoniae, Plasmodium species, and HIV) signifies that the administration of antimicrobial agents—once thought to be a panacea—requires appropriate stewardship. For all these reasons, infectious diseases continue to exert grim effects on individual patients as well as on international public health. Even with all the successes of the past century, physicians must be as thoughtful about infectious diseases now as they were at the beginning of the twentieth century.
GLOBAL CONSIDERATIONS
![]() Infectious diseases remain the second leading cause of death worldwide. Although the rate of infectious disease–related deaths has decreased dramatically over the past 20 years, the absolute numbers of such deaths have remained relatively constant, totaling just over 12 million in 2010 (Fig. 144-1A). As shown in Fig. 144-1B, these deaths disproportionately affect low- and middle-income countries (Chap. 13e); in 2010, 23% of all deaths worldwide were related to infectious diseases, with rates >60% in most sub-Saharan African countries.
Infectious diseases remain the second leading cause of death worldwide. Although the rate of infectious disease–related deaths has decreased dramatically over the past 20 years, the absolute numbers of such deaths have remained relatively constant, totaling just over 12 million in 2010 (Fig. 144-1A). As shown in Fig. 144-1B, these deaths disproportionately affect low- and middle-income countries (Chap. 13e); in 2010, 23% of all deaths worldwide were related to infectious diseases, with rates >60% in most sub-Saharan African countries.
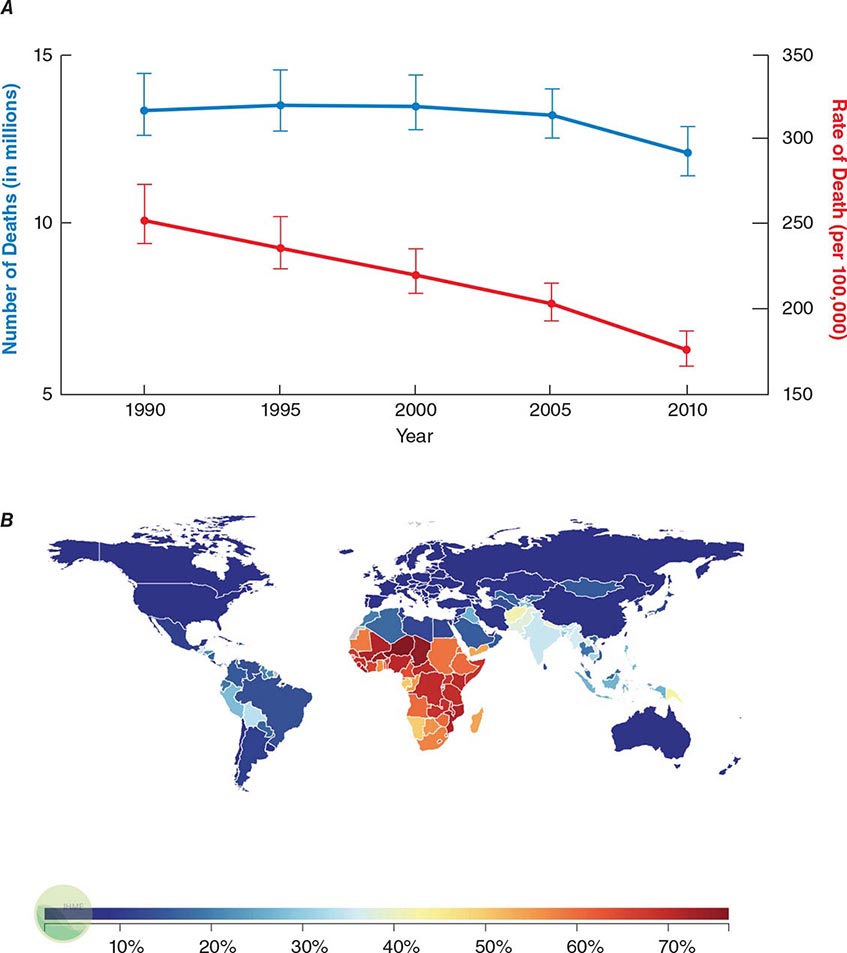
FIGURE 144-1 Magnitude of infectious disease–related deaths globally. A. The absolute number (blue line; left axis) and rate (red line; right axis) of infectious disease–related deaths throughout the world since 1990. B. A map depicting country-specific data for the percentages of total deaths that were attributable to communicable, maternal, neonatal, and nutritional disorders in 2010. (Source: Global Burden of Disease Study, Institute for Health Metrics and Evaluation.)
Given that infectious diseases are still a major cause of global mortality, understanding the local epidemiology of disease is critically important in evaluating patients. Diseases such as HIV/AIDS have decimated sub-Saharan Africa, with HIV-infected adults representing 15–26% of the total population in countries like Zimbabwe, Botswana, and Swaziland. Moreover, drug-resistant tuberculosis is rampant throughout the former Soviet-bloc countries, India, China, and South Africa. The ready availability of this type of information allows physicians to develop appropriate differential diagnoses and treatment plans for individual patients. Programs such as the Global Burden of Disease seek to quantify human losses (e.g., deaths, disability-adjusted life years) due to diseases by age, sex, and country over time; these data not only help inform local, national, and international health policy but can also help guide local medical decision-making. Even though some diseases (e.g., pandemic influenza, SARS) are seemingly geographically restricted, the increasing ease of rapid worldwide travel has raised concern about their swift spread around the globe. The world’s increasing interconnectedness has profound implications not only for the global economy but also for medicine and the spread of infectious diseases.
UNDERSTANDING THE MICROBIOTA
Normal, healthy humans are colonized with over 100 trillion bacteria as well as countless viruses, fungi, and archaea; taken together, these microorganisms outnumber human cells by 10–100 times (Chap. 86e). The major reservoir of these microbes is the gastrointestinal tract, but very substantial numbers of microbes live in the female genital tract, the oral cavity, and the nasopharynx. There is increasing interest in the skin and even the lungs as sites where microbial colonization might be highly relevant to the biology and disease susceptibility of the host. These commensal organisms provide the host with myriad benefits, from aiding in metabolism to shaping the immune system. With regard to infectious diseases, the vast majority of infections are caused by organisms that are part of the normal flora (e.g., S. aureus, S. pneumoniae, Pseudomonas aeruginosa), with relatively few infections due to organisms that are strictly pathogens (e.g, Neisseria gonorrhoeae, rabies virus). Perhaps it is not surprising that a general understanding of the microbiota is essential in the evaluation of infectious diseases. Individuals’ microbiotas likely have a major impact on their susceptibility to infectious diseases and even their responses to vaccines. Site-specific knowledge of the indigenous flora may facilitate appropriate interpretation of culture results, aid in selection of empirical antimicrobial therapy based on the likely causative agents, and provide additional impetus for rational antibiotic use to minimize the untoward effects of these drugs on the “beneficial” microbes that inhabit the body.
WHEN TO CONSIDER AN INFECTIOUS ETIOLOGY
The title of this chapter may appear to presuppose that the physician knows when a patient has an infectious disease. In reality, this chapter can serve only as a guide to the evaluation of a patient in whom an infectious disease is a possibility. Once a specific diagnosis is made, the reader should consult the subsequent chapters that deal with specific microorganisms in detail. The challenge for the physician is to recognize which patients may have an infectious disease as opposed to some other underlying disorder. This task is greatly complicated by the fact that infections have an infinite range of presentations, from acute life-threatening conditions (e.g., meningococcemia) to chronic diseases of varying severity (e.g., H. pylori–associated peptic ulcer disease) to no symptoms at all (e.g., latent M. tuberculosis infection). While it is impossible to generalize about a presentation that encompasses all infections, common findings in the history, physical examination, and basic laboratory testing often suggest that the patient either has an infectious disease or should be more closely evaluated for one. This chapter focuses on these common findings and how they may direct the ongoing evaluation of the patient.
APPROACH TO THE PATIENT:
Infectious Disease
See also Chap. 147.
HISTORY
As in all of medicine, obtaining a complete and thorough history is paramount in the evaluation of a patient with a possible infectious disease. The history is critical for developing a focused differential diagnosis and for guiding the physical exam and initial diagnostic testing. Although detailing all the elements of a history is beyond the scope of this chapter, specific components relevant to infectious diseases require particular attention. In general, these aspects focus on two areas: (1) an exposure history that may identify microorganisms with which the patient may have come into contact and (2) host-specific factors that may predispose to the development of an infection.
Exposure History • HISTORY OF INFECTIONS OR EXPOSURE TO DRUG-RESISTANT MICROBES Knowledge about a patient’s previous infections, with the associated microbial susceptibility profiles, is very helpful in determining possible etiologic agents. Specifically, knowing whether a patient has a history of infection with drug-resistant organisms (e.g., methicillin-resistant S. aureus, vancomycin-resistant Enterococcus species, enteric organisms that produce an extended-spectrum β-lactamase or carbapenemase) or may have been exposed to drug-resistant microbes (e.g., during a recent stay in a hospital, nursing home, or long-term acute-care facility) may alter the choice of empirical antibiotics. For example, a patient presenting with sepsis who is known to have a history of invasive infection with a multidrug-resistant isolate of P. aeruginosa should be treated empirically with an antimicrobial regimen that will cover this strain.
SOCIAL HISTORY Although the social history taken by physicians is often limited to inquiries about a patient’s alcohol and tobacco use, a complete social history can offer a number of clues to the underlying diagnosis. Knowing whether the patient has any high-risk behaviors (e.g., unsafe sexual behaviors, IV drug use), potential hobby-associated exposures (e.g., avid gardening, with possible Sporothrix schenckii exposure), or occupational exposures (e.g., increased risk for M. tuberculosis exposure in funeral service workers) can facilitate diagnosis. The importance of the social history is exemplified by a case in 2009 in which a laboratory researcher died of a Yersinia pestis infection acquired during his work; although this patient had visited both an outpatient clinic and an emergency department, his records at both sites failed to include his occupation—information that potentially could have led quickly to appropriate treatment and infection control measures.
DIETARY HABITS As certain pathogens are associated with specific dietary habits, inquiring about a patient’s diet can provide insight into possible exposures. For example, Shiga toxin–producing strains of Escherichia coli and Toxoplasma gondii are associated with the consumption of raw or undercooked meat; Salmonella typhimurium, Listeria monocytogenes, and Mycobacterium bovis with unpasteurized milk; Leptospira species, parasites, and enteric bacteria with unpurified water; and Vibrio species, norovirus, helminths, and protozoa with raw seafood.
ANIMAL EXPOSURES Because animals are often important vectors of infectious diseases, patients should be asked about exposures to any animals, including contact with their own pets, visits to petting zoos, or random encounters (e.g., home rodent infestation). For example, dogs can carry ticks that serve as agents for the transmission of several infectious diseases, including Lyme disease, Rocky Mountain spotted fever, and ehrlichiosis. Cats are associated with Bartonella henselae infection, reptiles with Salmonella infection, rodents with leptospirosis, and rabbits with tularemia (Chap. 167e).
TRAVEL HISTORY Attention should be paid to both international and domestic travel. Fever in a patient who has recently returned from abroad significantly broadens the differential diagnosis (Chap. 149); even a remote history of international travel may reflect patients’ exposure to infections with pathogens such as M. tuberculosis or Strongyloides stercoralis. Similarly, domestic travel may have exposed patients to pathogens that are not normally found in their local environment and therefore may not routinely be considered in the differential diagnosis. For example, a patient who has recently visited California or Martha’s Vineyard may have been exposed to Coccidioides immitis or Francisella tularensis, respectively. Beyond simply identifying locations that a patient may have visited, the physician needs to delve deeper to learn what kinds of activities and behaviors the patient engaged in during travel (e.g., the types of food and sources of water consumed, freshwater swimming, animal exposures) and whether the patient had the necessary immunizations and/or took the necessary prophylactic medications prior to travel; these additional exposures, which the patient may not think to report without specific prompting, are as important as exposures during a patient’s routine daily living.
HOST-SPECIFIC FACTORS Because many opportunistic infections (e.g., with Pneumocystis jirovecii, Aspergillus species, or JC virus) affect only immunocompromised patients, it is of vital importance to determine the immune status of the patient. Defects in the immune system may be due to an underlying disease (e.g., malignancy, HIV infection, malnutrition), a medication (e.g., chemotherapy, glucocorticoids, monoclonal antibodies to components of the immune system), a treatment modality (e.g., total body irradiation, splenectomy), or a primary immunodeficiency. The type of infection for which the patient is at increased risk varies with the specific type of immune defect (Chap. 375e). In concert with determining whether a patient is immunocompromised for any reason, the physician should review the immunization record to ensure that the patient is adequately protected against vaccine-preventable diseases (Chap. 148).
PHYSICAL EXAMINATION
Similar to the history, a thorough physical examination is crucial in evaluating patients with an infectious disease. Some elements of the physical exam (e.g., skin, lymphatics) that are often performed in a cursory manner as a result of the ever-increasing pace of medical practice may help identify the underlying diagnosis. Moreover, serial exams are critical since new findings may appear as the illness progresses. A description of all the elements of a physical exam is beyond the scope of this chapter, but the following components have particular relevance to infectious diseases.
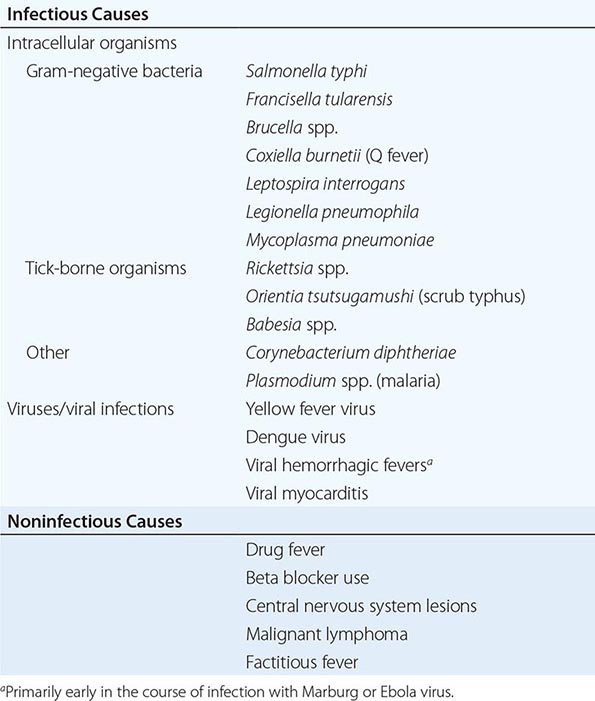
Vital Signs Given that elevations in temperature are often a hallmark of infection, paying close attention to the temperature may be of value in diagnosing an infectious disease. The idea that 37°C (98.6°F) is the normal human body temperature dates back to the nineteenth century and was initially based on axillary measurements. Rectal temperatures more accurately reflect the core body temperature and are 0.4°C (0.7°F) and 0.8°C (1.4°F) higher than oral and axillary temperatures, respectively. Although the definition of fever varies greatly throughout the medical literature, the most common definition, which is based on studies defining fever of unknown origin (Chap. 26), uses a temperature ≥38.3°C (101°F). Although fever is very commonly associated with infection, it is also documented in many other diseases (Chap. 23). For every 1°C (1.8°F) increase in core temperature, the heart rate typically rises by 15–20 beats/min. Table 144-1 lists infections that are associated with relative bradycardia (Faget’s sign), where patients have a lower heart rate than might be expected for a given body temperature. Although this pulse-temperature dissociation is not highly sensitive or specific for establishing a diagnosis, it is potentially useful in low-resource settings given its ready availability and simplicity.
CAUSES OF RELATIVE BRADYCARDIA |
Lymphatics There are ~600 lymph nodes throughout the body, and infections are an important cause of lymphadenopathy. A physical examination should include evaluation of lymph nodes in multiple regions (e.g., popliteal, inguinal, epitrochlear, axillary, multiple cervical regions), with notation of the location, size (normal, <1 cm), presence or absence of tenderness, and consistency (soft, firm, or shotty) and of whether the nodes are matted (i.e., connected and moving together). Of note, palpable epitrochlear nodes are always pathologic. Of patients presenting with lymphadenopathy, 75% have localized findings, and the remaining 25% have generalized lymphadenopathy (i.e., that involving more than one anatomic region). Localized lymphadenopathy in the head and neck region is found in 55% of patients, inguinal lymphadenopathy in 14%, and axillary lymphadenopathy in 5%. Determining whether the patient has generalized versus localized lymphadenopathy can help narrow the differential diagnosis, as various infections present differently.
Skin The fact that many infections have cutaneous manifestations gives the skin examination particular importance in the evaluation of patients (Chaps. 24, 25e, 72, and 156). It is important to perform a complete skin exam, with attention to both front and back. Specific rashes are often extremely helpful in narrowing the differential diagnosis of an infection (Chaps. 24 and 25e). In numerous anecdotal instances, patients in the intensive care unit have had “fever of unknown origin” that was actually due to unrecognized pressure ulcers. Moreover, close examination of the distal extremities for splinter hemorrhages, Janeway lesions, or Osler’s nodes may yield evidence of endocarditis or other causes of septic emboli.
Foreign Bodies As previously mentioned, many infections are caused by members of the indigenous microbiota. These infections typically occur when these microbes escape their normal habitat and enter a new one. Thus, maintenance of epithelial barriers is one of the most important mechanisms in protection against infection. However, hospitalization of patients is often associated with breaches of these barriers—e.g., due to placement of IV lines, surgical drains, or tubes (such as endotracheal tubes and Foley catheters) that allow microorganisms to localize in sites to which they normally would not have access (Chap. 168). Accordingly, knowing what lines, tubes, and drains are in place is helpful in ascertaining what body sites might be infected.
DIAGNOSTIC TESTING
Laboratory and radiologic testing has advanced greatly over the past few decades and has become an important component in the evaluation of patients. The dramatic increase in the number of serologic diagnostics, antigen tests, and molecular diagnostics available to the physician has, in fact, revolutionized medical care. However, all of these tests should be viewed as adjuncts to the history and physical examination—not a replacement for them. The selection of initial tests should be based directly on the patient’s history and physical exam findings. Moreover, diagnostic testing should generally be limited to those conditions that are reasonably likely and treatable, important in terms of public health considerations, and/or capable of providing a definitive diagnosis that will consequently limit other testing.
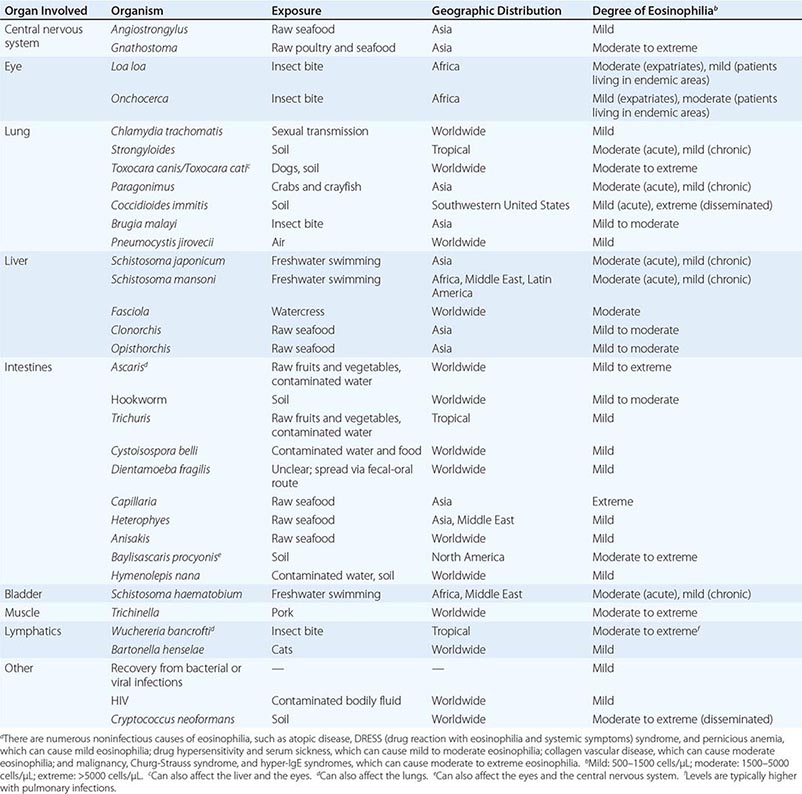
White Blood Cell (WBC) Count Elevations in the WBC count are often associated with infection, though many viral infections are associated with leukopenia. It is important to assess the WBC differential, given that different classes of microbes are associated with various leukocyte types. For example, bacteria are associated with an increase in polymorphonuclear neutrophils, often with elevated levels of earlier developmental forms such as bands; viruses are associated with an increase in lymphocytes; and certain parasites are associated with an increase in eosinophils. Table 144-2 lists the major infectious causes of eosinophilia.
MAJOR INFECTIOUS CAUSES OF EOSINOPHILIAa |
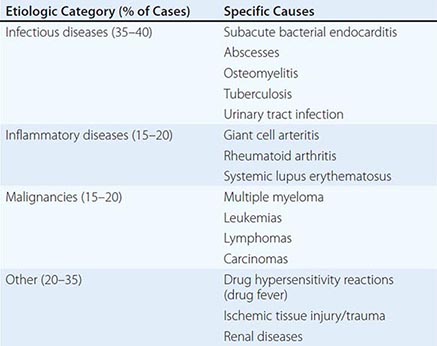
Inflammatory Markers The erythrocyte sedimentation rate (ESR) and the C-reactive protein (CRP) level are indirect and direct measures of the acute-phase response, respectively, that can be used to assess a patient’s general level of inflammation. Moreover, these markers can be followed serially over time to monitor disease progress/resolution. It is noteworthy that the ESR changes relatively slowly, and its measurement more often than weekly usually is not useful; in contrast, CRP concentrations change rapidly, and daily measurements can be useful in the appropriate context. Although these markers are sensitive indicators of inflammation, neither is very specific. An extremely elevated ESR (>100 mm/h) has a 90% predictive value for a serious underlying disease (Table 144-3). Work is ongoing to identify other potentially useful inflammatory markers (e.g., procalcitonin, serum amyloid A protein); however, their clinical utility requires further validation.
CAUSES OF AN EXTREMELY ELEVATED ERYTHROCYTE SEDIMENTATION RATE (>100 mm/h) |
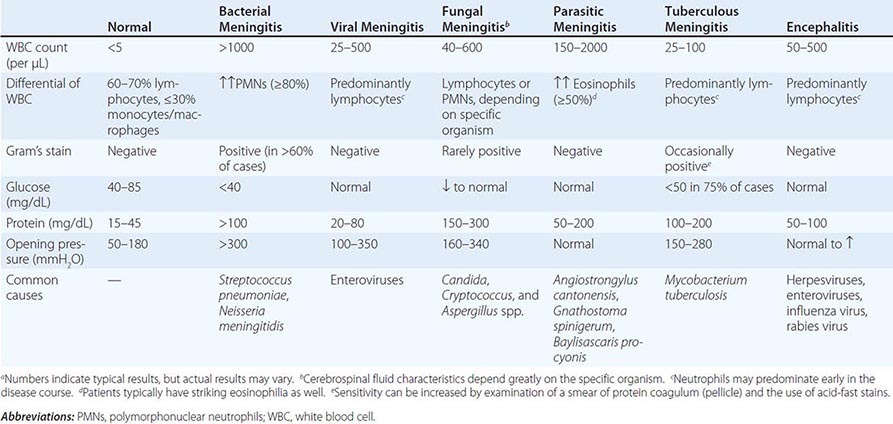
Analysis of Cerebrospinal Fluid (CSF) Assessment of CSF is critical for patients with suspected meningitis or encephalitis. An opening pressure should always be recorded, and fluid should routinely be sent for cell counts, Gram’s stain and culture, and determination of glucose and protein levels. A CSF Gram’s stain typically requires >105 bacteria/mL for reliable positivity; its specificity approaches 100%. Table 144-4 lists the typical CSF profiles for various infections. In general, CSF with a lymphocytic pleocytosis and a low glucose concentration suggests either infection (e.g., with Listeria, M. tuberculosis, or a fungus) or a noninfectious disorder (e.g, neoplastic meningitis, sarcoidosis). Bacterial antigen testing of CSF (e.g., latex agglutination tests for Haemophilus influenzae type b, group B Streptococcus, S. pneumoniae, and Neisseria meningitidis) is not recommended as a screening assay, given that these tests are no more sensitive than Gram’s stain; however, these assays can be helpful in presumptively identifying organisms seen on Gram’s stain. In contrast, other antigen tests (e.g., for Cryptococcus) and some CSF serologic testing (e.g., for Treponema pallidum, Coccidioides) are highly sensitive and are useful for select patients. In addition, polymerase chain reaction (PCR) analysis of CSF is increasingly being used for the diagnosis of bacterial (e.g., N. meningitidis, S. pneumoniae, mycobacteria) and viral (e.g., herpes simplex virus, enterovirus) infections; while these molecular tests permit rapid diagnosis with a high degree of sensitivity and specificity, they often do not allow determination of antimicrobial resistance profiles.
TYPICAL CSF PROFILES FOR MENINGITIS AND ENCEPHALITISa |
Cultures The mainstays of infectious disease diagnosis include the culture of infected tissue (e.g., surgical specimens) or fluid (e.g., blood, urine, sputum, purulence from a wound). Samples can be sent for culture of bacteria (aerobic or anaerobic), fungi, or viruses. Ideally, specimens are collected before the administration of antimicrobial therapy; in instances where this order of events is not clinically feasible, microscopic examination of the specimen (e.g., Gram-stained or potassium hydroxide [KOH]–treated preparations) is particularly important. Culture of the organism(s) allows identification of the etiologic agent, determination of the antimicrobial susceptibility profile, and—when there is concern about an outbreak—isolate typing. While cultures are extremely useful in the evaluation of patients, determining whether culture results are clinically meaningful or represent contamination (e.g., a non-aureus, non-lugdunensis staphylococcal species growing in a blood culture) can sometimes be challenging and requires an understanding of the patient’s immune status, exposure history, and microbiota. In some cases, serial cultures to demonstrate clearance of the organism may be helpful.
Pathogen-Specific Testing Numerous pathogen-specific tests (e.g., serology, antigen testing, PCR testing) are commercially available, and many hospitals now offer some of these tests in-house to facilitate rapid turnaround that ultimately enhances patient care. The reader is directed to relevant chapters on the pathogens of interest for specific details. Some of these tests (e.g., universal PCRs) identify organisms that currently are not cultivable and have unclear relationships to disease, thereby complicating diagnosis. As these tests become more commonplace and the work of the Human Microbiome Project progresses, the relevance of some of these previously unrecognized bacteria to human health will likely become more apparent.
Radiology Imaging provides an important adjunct to the physical examination, allowing evaluation for lymphadenopathy in regions that are not externally accessible (e.g., mediastinum, intraabdominal sites), assessment of internal organs for evidence of infection, and facilitation of image-guided percutaneous sampling of deep spaces. The choice of imaging modality (e.g., CT, MRI, ultrasound, nuclear medicine, use of contrast) is best made in consultation with a radiologist to ensure that the results will address the physician’s specific concerns.
TREATMENT
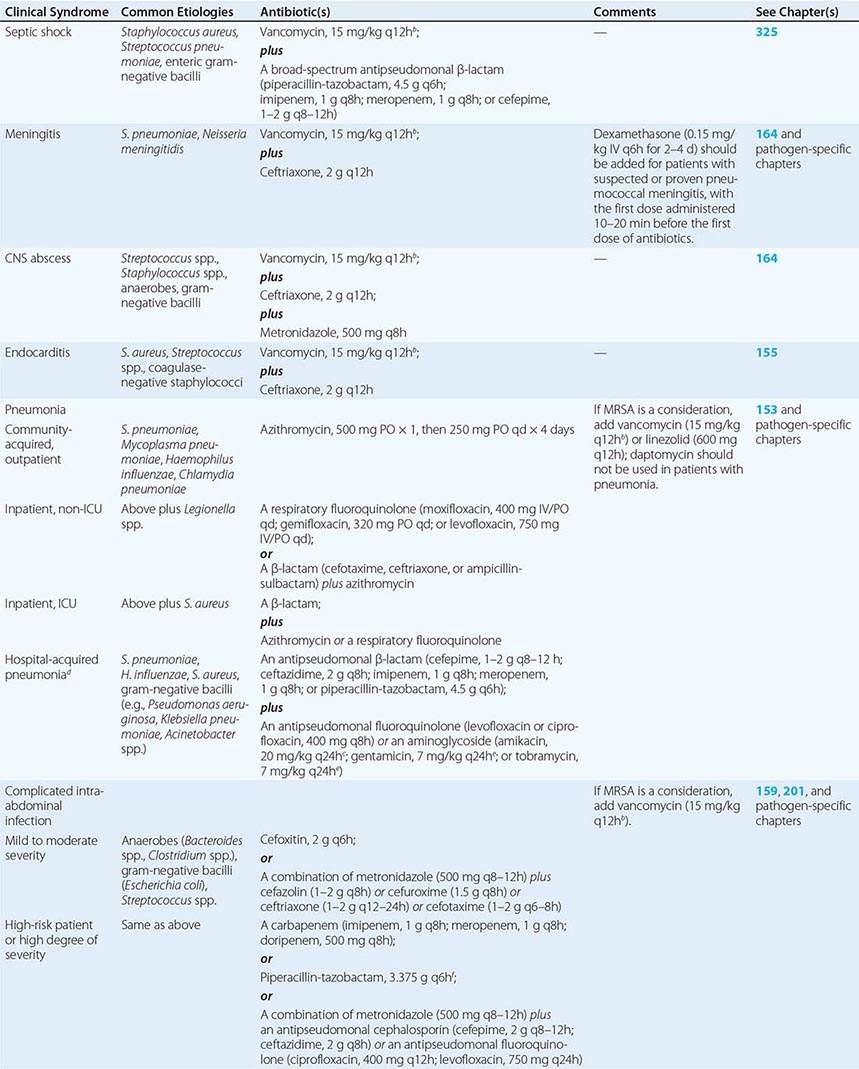
Physicians often must balance the need for empirical antibiotic treatment with the patient’s clinical condition. When clinically feasible, it is best to obtain relevant samples (e.g., blood, CSF, tissue, purulent exudate) for culture prior to the administration of antibiotics, as antibiotic treatment often makes subsequent diagnosis more difficult. Although a general maxim for antibiotic treatment is to use a regimen with as narrow a spectrum as possible (Chap. 170), empirical regimens are necessarily somewhat broad, given that a specific diagnosis has not yet been made. Table 144-5 lists empirical antibiotic treatment regimens for commonly encountered infectious presentations. These regimens should be narrowed as appropriate once a specific diagnosis is made. In addition to antibiotics, there is sometimes a role for adjunctive therapies, such as intravenous immunoglobulin G (IVIG) pooled from healthy adults or hyperimmune globulin prepared from the blood of individuals with high titers of specific antibodies to select pathogens (e.g., cytomegalovirus, hepatitis B virus, rabies virus, vaccinia virus, Clostridium tetani, varicella-zoster virus, Clostridium botulinum toxin). Although the data suggesting efficacy are limited, IVIG is often used for patients with suspected staphylococcal or streptococcal toxic shock syndrome.
INITIAL EMPIRICAL ANTIBIOTIC THERAPY FOR COMMON INFECTIOUS DISEASE PRESENTATIONSa |

INFECTION CONTROL
When evaluating a patient with a suspected infectious disease, the physician must consider what infection control methods are necessary to prevent transmission of any possible infection to other people. In 2007, the U.S. Centers for Disease Control and Prevention published guidelines for isolation precautions that are available for download at www.cdc.gov/hicpac/2007IP/2007isolationPrecautions.html. Persons exposed to certain pathogens (e.g., N. meningitidis, HIV, Bacillus anthracis) should receive postexposure prophylaxis to prevent disease acquisition. (See relevant chapters for details on specific pathogens.)
WHEN TO OBTAIN AN INFECTIOUS DISEASE CONSULT
At times, primary physicians need assistance with patient management, from a diagnostic and/or therapeutic perspective. Multiple studies have demonstrated that an infectious disease consult is associated with positive outcomes for patients with various diseases. For example, in a prospective cohort study of patients with S. aureus bacteremia, infectious disease consultation was independently associated with a 56% reduction in 28-day mortality. In addition, infectious disease specialists provide other services (e.g., infection control, antimicrobial stewardship, management of outpatient antibiotic therapy, occupational exposure programs) that have been shown to benefit patients. Whenever such assistance would be advantageous to a patient with a possible infection, the primary physician should opt for an infectious disease consult. Specific situations that might prompt a consult include (1) difficult-to-diagnose patients with presumed infections, (2) patients who are not responding to treatment as expected, (3) patients with a complicated medical history (e.g., organ transplant recipients, patients immunosuppressed due to autoimmune or inflammatory conditions), and (4) patients with “exotic” diseases (i.e., diseases that are not typically seen within the region).
PERSPECTIVE
The study of infectious diseases is really a study of host-bacterial interactions and represents evolution by both the host and the bacteria—an endless struggle in which microbes have generally been more creative and adaptive. Given that nearly one-quarter of deaths worldwide are still related to infectious diseases, it is clear that the war against infectious diseases has not been won. For example, a cure for HIV infection is still lacking, there have been only marginal improvements in the methods for detection and treatment of tuberculosis after more than a half century of research, new infectious diseases (e.g., pandemic influenza, viral hemorrhagic fevers) continue to emerge, and the threat of microbial bioterrorism remains high. The subsequent chapters in Part 8 detail—on both a syndrome and a microbe-by-microbe basis—the current state of medical knowledge about infectious diseases. At their core, all of these chapters carry a similar message: Despite numerous advances in the diagnosis, treatment, and prevention of infectious diseases, much work and research are required before anyone can confidently claim that “all the major infections have disappeared.” In reality, this goal will never be attained, given the rapid adaptability of microbes.
145e | Molecular Mechanisms of Microbial Pathogenesis |
Over the past four decades, molecular studies of the pathogenesis of microorganisms have yielded an explosion of information about the various microbial and host molecules that contribute to the processes of infection and disease. These processes can be classified into several stages: microbial encounter with and entry into the host; microbial growth after entry; avoidance of innate host defenses; tissue invasion and tropism; tissue damage; and transmission to new hosts. Virulence is the measure of an organism’s capacity to cause disease and is a function of the pathogenic factors elaborated by microbes. These factors promote colonization (the simple presence of potentially pathogenic microbes in or on a host), infection (attachment and growth of pathogens and avoidance of host defenses), and disease (often, but not always, the result of activities of secreted toxins or toxic metabolites). In addition, the host’s inflammatory response to infection greatly contributes to disease and its attendant clinical signs and symptoms. The recent surge of interest in the role of the microbiota and its associated microbiome—the collection of microbial genomes residing in or on mammalian organisms—in the physiology of, susceptibility to, and response to infection and in immune system development has had an enormous impact on our understanding of host-pathogen interaction.
THE MICROBIOME
(See also Chap. 86e) We now understand that the indigenous microbial organisms living in close association with almost all animals are organized into complex communities that strongly modulate the ability of pathogenic microbes to become established in or on host surfaces. The sheer numbers of these microbes and their genomic variability vastly exceed the numbers of host cells and genes in a typical animal. Changes and differences in microbiomes within and between individuals, currently characterized by high-throughput DNA sequencing techniques and bioinformatic analysis, affect the development and control of the immune system as well as such diverse conditions as obesity, type 1 diabetes, cognition, neurologic states, autoimmune diseases, and infectious diseases of the skin, gastrointestinal tract, respiratory tract, and vagina. It has been more difficult to directly associate specific types of microbiomes with pathophysiologic states and to assess how conserved or variable microbial species within human and animal microbiomes are evolving. Defining clusters of organisms associated with diseases may become more feasible as more data are obtained. Complicating this task are the results from the Human Microbiome Project suggesting a high level of variability among individuals in the components of the microbiome, although many individuals appear to maintain a fairly conserved microbiome throughout their lives. In the context of infectious diseases, clear changes and disruptions of the indigenous microbiome have a strong and often fundamental impact on the progression of infection. Such alterations can be associated with the effects of antibiotic and immunosuppressive drug use on the normal flora, with environmental changes, and with the impact of microbial virulence factors that displace the indigenous microbial flora to facilitate pathogen colonization. As the available technology for defining the microbiome expands, there is no doubt that the resulting data will markedly affect our concepts of and approaches to microbial pathogenesis and infectious disease treatment.
MICROBIAL ENTRY AND ADHERENCE
Entry Sites A microbial pathogen can potentially enter any part of a host organism. In general, the type of disease produced by a particular microbe is often a direct consequence of its route of entry into the body. The most common sites of entry are mucosal surfaces (the respiratory, alimentary, and urogenital tracts) and the skin. Ingestion, inhalation, and sexual contact are typical routes of microbial entry. Other portals of entry include sites of skin injury (cuts, bites, burns, trauma) along with injection via natural (i.e., vector-borne) or artificial (i.e., needle-stick injury) routes. A few pathogens, such as Schistosoma species, can penetrate unbroken skin. The conjunctiva can serve as an entry point for pathogens of the eye, which occasionally spread systemically from that site.
Microbial entry usually relies on the presence of specific factors needed for persistence and growth in a tissue. Fecal-oral spread via the alimentary tract requires a biologic profile consistent with survival in the varied environments of the gastrointestinal tract (including the low pH of the stomach and the high bile content of the intestine) as well as in contaminated food or water outside the host. Organisms that gain entry via the respiratory tract survive well in small moist droplets produced during sneezing and coughing. Pathogens that enter by venereal routes often survive best in the warm moist environment of the urogenital mucosa and have restricted host ranges (e.g., Neisseria gonorrhoeae, Treponema pallidum, and HIV).
The biology of microbes entering through the skin is highly varied. Some of these organisms can survive in a broad range of environments, such as the salivary glands or alimentary tracts of arthropod vectors, the mouths of larger animals, soil, and water. A complex biology allows protozoan parasites such as Plasmodium, Leishmania, and Trypanosoma species to undergo morphogenic changes that permit transmission to mammalian hosts during insect feeding for blood meals. Plasmodia are injected as infective sporozoites from the salivary glands during mosquito feeding. Leishmania parasites are regurgitated as promastigotes from the alimentary tract of sandflies and injected by bite into a susceptible host. Trypanosomes are first ingested from infected hosts by reduviid bugs; the pathogens then multiply in the gastrointestinal tract of the insects and are released in feces onto the host’s skin during subsequent feedings. Most microbes that land directly on intact skin are destined to die, as survival on the skin or in hair follicles requires resistance to fatty acids, low pH, and other antimicrobial factors on the skin. Once it is damaged (and particularly if it becomes necrotic), the skin can be a major portal of entry and growth for pathogens and elaboration of their toxic products. Burn wound infections and tetanus are clear examples. After animal bites, pathogens resident in the animal’s saliva gain access to the victim’s tissues through the damaged skin. Rabies is the paradigm for this pathogenic process; rabies virus grows in striated muscle cells at the site of inoculation.
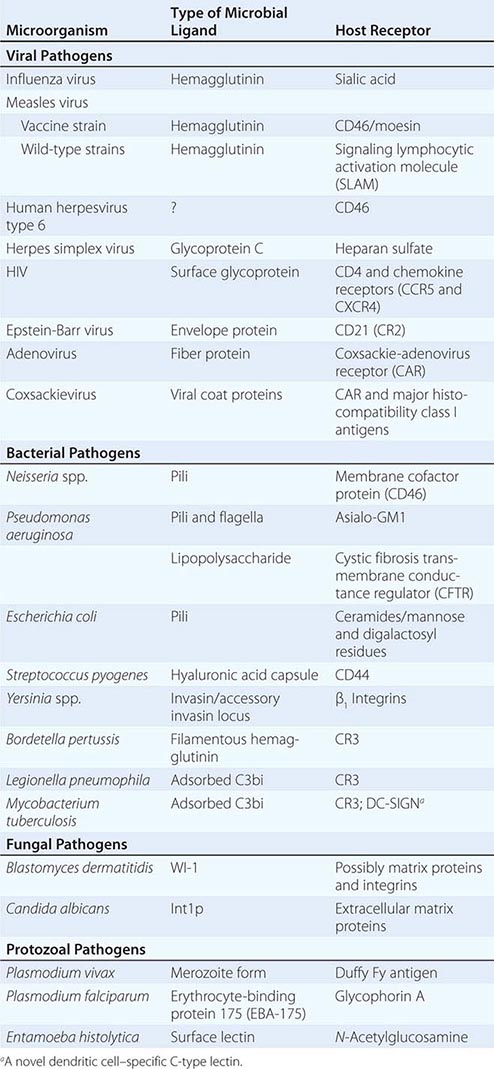
Microbial Adherence Once in or on a host, most microbes must anchor themselves to a tissue or tissue factor; the possible exceptions are organisms that directly enter the bloodstream and multiply there. Specific ligands or adhesins for host receptors constitute a major area of study in the field of microbial pathogenesis. Adhesins comprise a wide range of surface structures, not only anchoring the microbe to a tissue and promoting cellular entry where appropriate but also eliciting host responses critical to the pathogenic process (Table 145e-1). Most microbes produce multiple adhesins specific for multiple host receptors. These adhesins are often redundant, are serologically variable, and act additively or synergistically with other microbial factors to promote microbial sticking to host tissues. In addition, some microbes adsorb host proteins onto their surface and utilize the natural host protein receptor for microbial binding and entry into target cells.
EXAMPLES OF MICROBIAL LIGAND-RECEPTOR INTERACTIONS |
VIRAL ADHESINS All viral pathogens must bind to host cells, enter them, and replicate within them. Viral coat proteins serve as the ligands for cellular entry, and more than one ligand-receptor interaction may be needed; for example, HIV utilizes its envelope glycoprotein (gp) 120 to enter host cells by binding both to CD4 and to one of two receptors for chemokines (designated CCR5 and CXCR4). Similarly, the measles virus H glycoprotein binds to both CD46 and the membrane-organizing protein moesin on host cells. The gB and gC proteins on herpes simplex virus bind to heparan sulfate, although this adherence is not essential for entry but rather serves to concentrate virions close to the cell surface; this step is followed by attachment to mammalian cells mediated by the viral gD protein, with subsequent formation of a homotrimer of viral gB protein or a heterodimer of viral gH and gL proteins that permits fusion of the viral envelope with the host cell membrane. Herpes simplex virus can use a number of eukaryotic cell surface receptors for entry, including the herpesvirus entry mediator (related to the tumor necrosis factor receptor), members of the immunoglobulin superfamily, the proteins nectin-1 and nectin-2, and modified heparan sulfate.
BACTERIAL ADHESINS Among the microbial adhesins studied in greatest detail are bacterial pili and flagella (Fig. 145e-1). Pili or fimbriae are commonly used by gram-negative bacteria for attachment to host cells and tissues; studies have identified similar factors produced by gram-positive organisms such as group B streptococci. In electron micrographs, these hairlike projections (up to several hundred per cell) may be confined to one end of the organism (polar pili) or distributed more evenly over the surface. An individual cell may have pili with a variety of functions. Most pili are made up of a major pilin protein subunit (molecular weight, 17,000–30,000) that polymerizes to form the pilus. Many strains of Escherichia coli isolated from urinary tract infections express mannose-binding type 1 pili, whose binding to integral membrane glycoproteins called uroplakins that coat the cells in the bladder epithelium is inhibited by D-mannose. Other strains produce the Pap (pyelonephritis-associated) or P pilus adhesin that mediates binding to digalactose (gal-gal) residues on globosides of the human P blood groups. Both of these types of pili have proteins located at the tips of the main pilus unit that are critical to the binding specificity of the whole pilus unit. Although immunization with the mannose-binding tip protein (FimH) of type 1 pili prevents experimental E. coli bladder infections in mice and monkeys, a human trial of this vaccine was not successful. E. coli cells causing diarrheal disease express pilus-like receptors for enterocytes on the small bowel, along with other receptors termed colonization factors.
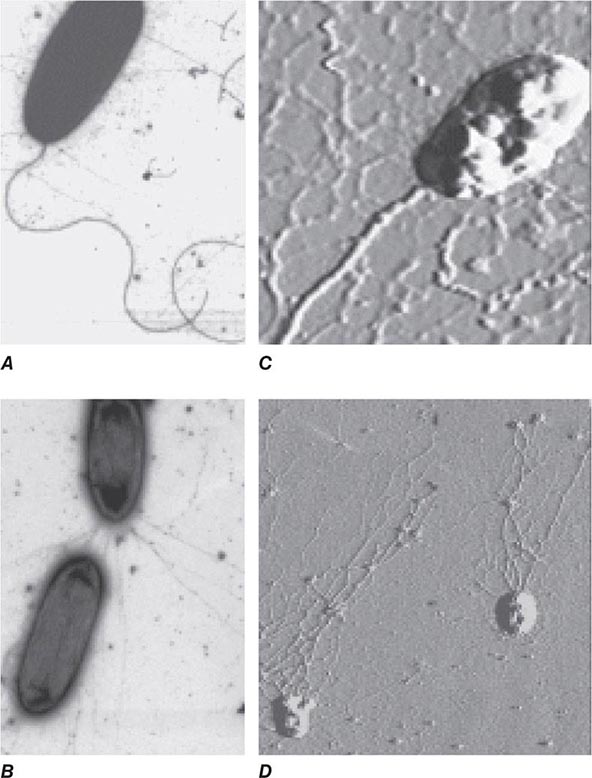
FIGURE 145e-1 Bacterial surface structures. A and B. Traditional electron micrographic images of fixed cells of Pseudomonas aeruginosa. Flagella (A) and pili (B) project out from the bacterial poles. C and D. Atomic force microscopic image of live P. aeruginosa freshly planted onto a smooth mica surface. This technology reveals the fine, three-dimensional detail of the bacterial surface structures. (Images courtesy of Drs. Martin Lee and Milan Bajmoczi, Harvard Medical School.)
The type IV pilus, a common type of pilus found in Neisseria species, Moraxella species, Vibrio cholerae, Legionella pneumophila, Salmonella enterica serovar Typhi, enteropathogenic E. coli, and Pseudomonas aeruginosa, often mediates adherence of organisms to target surfaces. Type IV pili tend to have a relatively conserved aminoterminal region and a more variable carboxyl-terminal region. For some species (e.g., N. gonorrhoeae, Neisseria meningitidis, and enteropathogenic E. coli), the pili are critical for attachment to mucosal epithelial cells. For others, such as P. aeruginosa, the pili only partially mediate the cells’ adherence to host tissues and may in some circumstances inhibit colonization. For example, a recent study of P. aeruginosa colonization of the gastrointestinal tract of mice evaluated a bank of mutants in which all nonessential genes were interrupted; those mutants that were unable to produce the type IVa pili were actually better able to colonize the gastrointestinal mucosa, although the basis for this observation was not identified. V. cholerae cells appear to use two different types of pili for intestinal colonization. Whereas interference with this stage of colonization would appear to be an effective antibacterial strategy, attempts to develop pilus-based vaccines for human diseases have not been highly successful to date.
Flagella are long appendages attached at either one or both ends of the bacterial cell (polar flagella) or distributed over the entire cell surface (peritrichous flagella). Flagella, like pili, are composed of a polymerized or aggregated basic protein. In flagella, the protein subunits form a tight helical structure and vary serologically with the species. Spirochetes such as T. pallidum and Borrelia burgdorferi have axial filaments similar to flagella running down the long axis of the center of the cell, and they “swim” by rotation around these filaments. Some bacteria can glide over a surface in the absence of obvious motility structures.
Other bacterial structures involved in adherence to host tissues include specific staphylococcal and streptococcal proteins that bind to human extracellular matrix proteins such as fibrin, fibronectin, fibrinogen, laminin, and collagen. Fibronectin appears to be a commonly used receptor for various pathogens; a particular amino acid sequence in fibronectin, Arg-Gly-Asp or RGD, is a critical target used by bacteria to bind to host tissues. Binding of a highly conserved Staphylococcus aureus surface protein, clumping factor A (ClfA), to fibrinogen has been implicated in many aspects of pathogenesis. Attempts to interrupt this interaction and prevent S. aureus sepsis in low-birth-weight infants by administering an intravenous IgG preparation derived from the plasma of individuals with high titers of antibody to ClfA failed to show efficacy in a clinical trial; however, this approach is being pursued in some vaccine formulations targeting this organism. The conserved outer-core portion of the lipopolysaccharide (LPS) of P. aeruginosa mediates binding to the cystic fibrosis transmembrane conductance regulator (CFTR) on airway epithelial cells—an event that appears to play a critical role in normal host resistance to infection by initiating recruitment of polymorphonuclear neutrophils (PMNs) to the lung mucosa to kill the cells via opsonophagocytosis. A large number of microbial pathogens encompassing major gram-positive bacteria (staphylococci and streptococci), gram-negative bacteria (major enteric species and coccobacilli), fungi (Candida, Fusobacterium, Aspergillus), and even eukaryotes (Trichomonas vaginalis and Plasmodium falciparum) express a surface polysaccharide composed of β-1-6-linked-poly-N-acetyl-D-glucosamine (PNAG). One of the functions of PNAG for some of these organisms is to promote binding to materials used in catheters and other types of implanted devices. This polysaccharide may be a critical factor in the establishment of device-related infections by pathogens such as staphylococci and E. coli. High-powered imaging techniques (e.g., atomic force microscopy) have revealed that bacterial cells have a nonhomogeneous surface that is probably attributable to different concentrations of cell surface molecules, including microbial adhesins, at specific places on the cell surface (Figs. 120-1C and 120-1D).
FUNGAL ADHESINS Several fungal adhesins have been described that mediate colonization of epithelial surfaces, particularly adherence to structures like fibronectin, laminin, and collagen. The product of the Candida albicans INT1 gene, Int1p, bears similarity to mammalian integrins that bind to extracellular matrix proteins. The agglutinin-like sequence (ALS) adhesins are large cell-surface glycoproteins mediating adherence of pathogenic Candida to host tissues. These adhesins possess a conserved three-domain structure composed of an N-terminal domain that mediates adherence to host tissue receptors, a central motif consisting of a number of repeats of a conserved sequence of 36 amino acids, and a C-terminal domain that varies in length and sequence and contains a glycosylphosphatidylinositol (GPI) anchor addition site that allows binding of the adhesin to the fungal cell wall. Variability in the number of central domains in different ALS proteins characterizes different adhesins with specificity for different host receptors. The ALS adhesins are expressed under certain environmental conditions and are crucial for pathogenesis of fungal infections.
For several fungal pathogens that initiate infections after inhalation of infectious material, the inoculum is ingested by alveolar macrophages, in which the fungal cells transform to pathogenic phenotypes. Like C. albicans, Blastomyces dermatitidis binds to CD11b/CD18 integrins as well as to CD14 on macrophages. B. dermatitidis produces a 120-kDa surface protein, designated WI-1, that mediates this adherence. An unidentified factor on Histoplasma capsulatum also mediates binding of this fungal pathogen to the integrin surface proteins.
EUKARYOTIC PATHOGEN ADHESINS Eukaryotic parasites use complicated surface glycoproteins as adhesins, some of which are lectins (proteins that bind to specific carbohydrates on host cells). For example, Plasmodium vivax, one of six Plasmodium species causing malaria, binds (via Duffy-binding protein) to the Duffy blood group carbohydrate antigen Fy on erythrocytes. Entamoeba histolytica, the third leading cause of death from parasitic diseases, expresses two proteins that bind to the disaccharide galactose/N-acetyl galactosamine. Reports indicate that children with mucosal IgA antibody to one of these lectins are resistant to reinfection with virulent E. histolytica. A major surface glycoprotein (gp63) of Leishmania promastigotes is needed for these parasites to enter human macrophages—the principal target cell of infection. This glycoprotein promotes complement binding but inhibits complement lytic activity, allowing the parasite to use complement receptors for entry into macrophages; gp63 also binds to fibronectin receptors on macrophages. In addition, the pathogen can express a carbohydrate that mediates binding to host cells. Evidence suggests that, as part of hepatic granuloma formation, Schistosoma mansoni expresses a carbohydrate epitope related to the Lewis × blood group antigen that promotes adherence of helminthic eggs to vascular endothelial cells under inflammatory conditions.
Host Receptors Host receptors are found both on target cells (such as epithelial cells lining mucosal surfaces) and within the mucus layer covering these cells. Microbial pathogens bind to a wide range of host receptors to establish infection (Table 145e-1). Selective loss of host receptors for a pathogen may confer natural resistance to an otherwise susceptible population. For example, 70% of individuals in West Africa lack Fy antigens and are resistant to P. vivax infection. S. enterica serovar Typhi, the etiologic agent of typhoid fever, produces a pilus protein that binds to CFTR to enter the gastrointestinal submucosa after being ingested by enterocytes. As homozygous mutations in CFTR are the cause of the life-shortening disease cystic fibrosis, heterozygote carriers (e.g., 4–5% of individuals of European ancestry) may have had a selective advantage due to decreased susceptibility to typhoid fever.
Numerous virus–target cell interactions have been described, and it is now clear that different viruses can use similar host cell receptors for entry. The list of certain and likely host receptors for viral pathogens is long. Among the host membrane components that can serve as receptors for viruses are sialic acids, gangliosides, glycosaminoglycans, integrins and other members of the immunoglobulin superfamily, histocompatibility antigens, and regulators and receptors for complement components. A notable example of the effect of host receptors on the pathogenesis of infection has emerged from studies comparing the binding of avian influenza A subtype H5N1 with that of influenza A strains expressing the H1 subtype of hemagglutinin. The H1 subtypes tend to be highly pathogenic and transmissible from human to human, and they bind to a receptor composed of two sugar molecules: sialic acid linked α-2-6 to galactose. This receptor is expressed at high levels in the airway epithelium; when virus is shed from this surface, its transmission via coughing and aerosol droplets is facilitated. In contrast, the H5N1 avian influenza virus binds to sialic acid linked α-2-3 to galactose, and this receptor is expressed at high levels in pneumocytes in the alveoli. Infection in the alveoli is thought to underlie the high mortality rate associated with avian influenza but also the low interhuman transmissibility of this strain, which is not readily transported to the airways from which it can be expelled by coughing. Nonetheless, it was recently shown that H5 hemagglutinins can acquire mutations that vastly increase their transmissibility while not affecting their high level of lethality.
MICROBIAL GROWTH AFTER ENTRY
Once established on a mucosal or skin site, pathogenic microbes must replicate before causing full-blown infection and disease. Within cells, viral particles release their nucleic acids, which may be directly translated into viral proteins (positive-strand RNA viruses), transcribed from a negative strand of RNA into a complementary mRNA (negative-strand RNA viruses), or transcribed into a complementary strand of DNA (retroviruses); for DNA viruses, mRNA may be transcribed directly from viral DNA, either in the cell nucleus or in the cytoplasm. To grow, bacteria must acquire specific nutrients or synthesize them from precursors in host tissues. Many infectious processes are usually confined to specific epithelial surfaces—e.g., H1 subtype influenza to the respiratory mucosa, gonorrhea to the urogenital epithelium, shigellosis to the gastrointestinal epithelium. While there are multiple reasons for this specificity, one important consideration is the ability of these pathogens to obtain from these specific environments the nutrients needed for growth and survival.
Temperature restrictions also play a role in limiting certain pathogens to specific tissues. Rhinoviruses, a cause of the common cold, grow best at 33°C and replicate in cooler nasal tissues but not in the lung. Leprosy lesions due to Mycobacterium leprae are found in and on relatively cool body sites. Fungal pathogens that infect the skin, hair follicles, and nails (dermatophyte infections) remain confined to the cooler, exterior, keratinous layer of the epithelium.
A topic of major interest is the ability of many bacterial, fungal, and protozoal species to grow in multicellular masses referred to as biofilms. These masses are biochemically and morphologically quite distinct from the free-living individual cells referred to as planktonic cells. Growth in biofilms leads to altered microbial metabolism, production of extracellular virulence factors, and decreased susceptibility to biocides, antimicrobial agents, and host defense molecules and cells. P. aeruginosa growing on the bronchial mucosa during chronic infection, staphylococci and other pathogens growing on implanted medical devices, and dental pathogens growing on tooth surfaces to form plaque are several examples of microbial biofilm growth associated with human disease. Many other pathogens can form biofilms during in vitro growth. It is increasingly accepted that this mode of growth contributes to microbial virulence and induction of disease and that biofilm formation can also be an important factor in microbial survival outside the host, promoting transmission to additional susceptible individuals.
AVOIDANCE OF INNATE HOST DEFENSES
As microbes have interacted with mucosal/epithelial surfaces since the emergence of multicellular organisms, it is not surprising that multicellular hosts have a variety of innate surface defense mechanisms that can sense when pathogens are present and contribute to their elimination. The skin is acidic and is bathed with fatty acids toxic to many microbes. Skin pathogens such as staphylococci must tolerate these adverse conditions. Mucosal surfaces are covered by a barrier composed of a thick mucus layer that entraps microbes and facilitates their transport out of the body by such processes as mucociliary clearance, coughing, and urination. Mucous secretions, saliva, and tears contain antibacterial factors such as lysozyme and antimicrobial peptides as well as antiviral factors such as interferons (IFNs). Gastric acidity and bile salts are inimical to the survival of many ingested pathogens, and most mucosal surfaces—particularly the nasopharynx, the vaginal tract, and the gastrointestinal tract—contain a resident flora of commensal microbes that interfere with the ability of pathogens to colonize and infect a host. Major advances in the use of nucleic acid sequencing now allow extensive identification and characterization of the vast array of commensal organisms that have come to be referred to as the microbiota. In addition to its role in providing competition for mucosal colonization, acquisition of a normal microbiota is critical for proper development of the immune system, influencing maturation and differentiation of components of both the innate and acquired arms.
Pathogens that survive local antimicrobial factors must still contend with host endocytic, phagocytic, and inflammatory responses as well as with host genetic factors that determine the degree to which a pathogen can survive and grow. The list of genes whose variants, usually by single-nucleotide polymorphisms, can affect host susceptibility and resistance to infection is rapidly expanding. A classic example is a 32-bp deletion in the gene for the HIV-1 co-receptor known as chemokine receptor 5 (CCR5), which, when present in the homozygous state, confers high-level resistance to HIV-1 infection. The growth of viral pathogens entering skin or mucosal epithelial cells can be limited by a variety of host genetic factors, including production of IFNs, modulation of receptors for viral entry, and age- and hormone-related susceptibility factors; by nutritional status; and even by personal habits such as smoking and exercise.
Encounters with Epithelial Cells Over the past two decades, many pathogens have been shown to enter epithelial cells (Fig. 145e-2); they often use specialized surface structures that bind to receptors, with consequent internalization. However, the exact role and the importance of this process in infection and disease are not well defined for most of these pathogens. Microbial entry into host epithelial cells is seen as a means for dissemination to adjacent or deeper tissues or as a route to sanctuary to avoid ingestion and killing by professional phagocytes. Epithelial cell entry appears, for instance, to be a critical aspect of dysentery induction by Shigella.
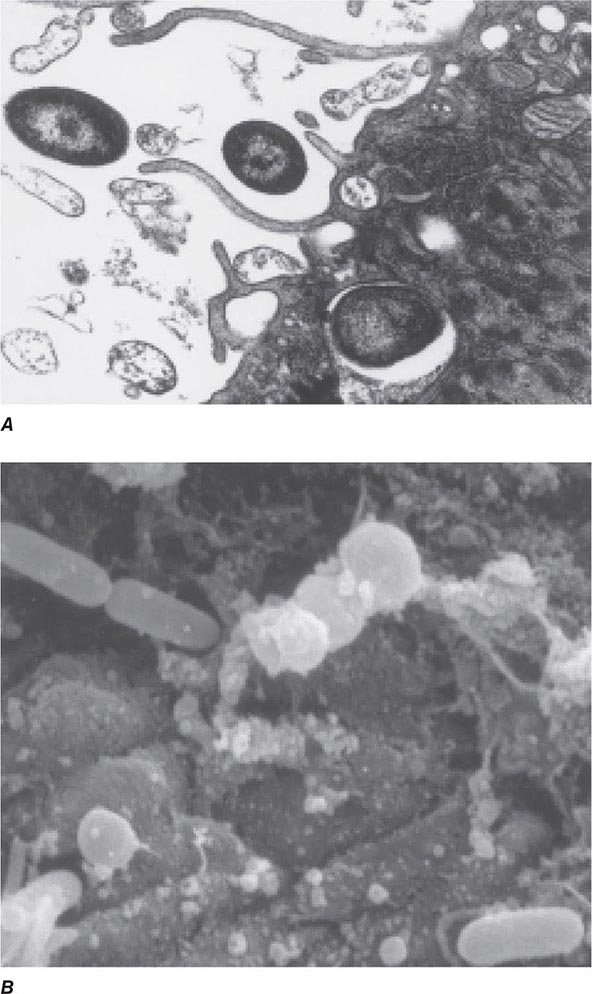
FIGURE 145e-2 Entry of bacteria into epithelial cells. A. Internalization of Pseudomonas aeruginosa by cultured airway epithelial cells expressing wild-type cystic fibrosis transmembrane conductance regulator, the cell receptor for bacterial ingestion. B. Entry of P. aeruginosa into murine tracheal epithelial cells after murine infection by the intranasal route.
Curiously, the less virulent strains of many bacterial pathogens are more adept at entering epithelial cells than are more virulent strains; examples include pathogens that lack the surface polysaccharide capsule needed to cause serious disease. Thus, for Haemophilus influenzae, Streptococcus pneumoniae, Streptococcus agalactiae (group B Streptococcus), and Streptococcus pyogenes, isogenic mutants or variants lacking capsules enter epithelial cells better than the wild-type, encapsulated parental forms that cause disseminated disease. These observations have led to the proposal that epithelial cell entry may be primarily a manifestation of host defense, resulting in bacterial clearance by both shedding of epithelial cells containing internalized bacteria and initiation of a protective and nonpathogenic inflammatory response. However, a possible consequence of this process could be the opening of a hole in the epithelium, potentially allowing uningested organisms to enter the submucosa. This scenario has been documented in murine S. enterica serovar Typhimurium infections and in experimental bladder infections with uropathogenic E. coli. In the latter system, bacterial pilus-mediated attachment to uroplakins induces exfoliation of the cells with attached bacteria. Subsequently, infection is produced by residual bacterial cells that invade the superficial bladder epithelium, where they can grow intracellularly into biofilm-like masses encased in an extracellular polysaccharide-rich matrix and surrounded by uroplakin. This mode of growth produces structures that have been referred to as bacterial pods. It is likely that at low bacterial inocula epithelial cell ingestion and subclinical inflammation are efficient means to eliminate pathogens, while at higher inocula a proportion of surviving bacterial cells enter the host tissue through the damaged mucosal surface and multiply, producing disease. Alternatively, failure of the appropriate epithelial cell response to a pathogen may allow the organism to survive on a mucosal surface where, if it avoids other host defenses, it can grow and cause a local infection. Along these lines, as noted above, P. aeruginosa is taken into epithelial cells by CFTR, a protein missing or nonfunctional in most severe cases of cystic fibrosis. The major clinical consequence of this disease is chronic airway-surface infection with P. aeruginosa in 80–90% of patients. The failure of airway epithelial cells to ingest and promote the removal of P. aeruginosa via a properly regulated inflammatory response has been proposed as a key component of the hypersusceptibility of cystic fibrosis patients to chronic airway infection with this organism.
Encounters with Phagocytes • PHAGOCYTOSIS AND INFLAMMATION Phagocytosis of microbes is a major innate host defense that limits the growth and spread of pathogens. Phagocytes appear rapidly at sites of infection in conjunction with the initiation of inflammation. Ingestion of microbes by both tissue-fixed macrophages and migrating phagocytes probably accounts for the limited ability of most microbial agents to cause disease. A family of related molecules called collectins, soluble defense collagens, or pattern-recognition molecules are found in blood (mannose-binding lectins), in lung (surfactant proteins A and D), and most likely in other tissues as well and bind to carbohydrates on microbial surfaces to promote phagocyte clearance. Bacterial pathogens seem to be ingested principally by PMNs, while eosinophils are frequently found at sites of infection by protozoan or multicellular parasites. Successful pathogens, by definition, must avoid being cleared by professional phagocytes. One of several antiphagocytic strategies employed by bacteria and by the fungal pathogen Cryptococcus neoformans is to elaborate large-molecular-weight surface polysaccharide antigens, often in the form of a capsule that coats the cell surface. Most pathogenic bacteria produce such antiphagocytic capsules. On occasion, proteins or polypeptides form capsule-like coatings for organisms such as group A streptococci and Bacillus anthracis.
As activation of local phagocytes in tissues is a key step in initiating inflammation and migration of additional phagocytes into infected sites, much attention has been paid to microbial factors that initiate inflammation. These are usually conserved factors critical to the microbes’ survival and are referred to as pathogen-associated molecular patterns (PAMPs). Cellular responses to microbial encounters with phagocytes are governed largely by the structure of the microbial PAMPs that elicit inflammation, and detailed knowledge of these structures of bacterial pathogens has contributed greatly to our understanding of molecular mechanisms of microbial pathogenesis mediated by activation of host cell molecules such as TLRs (Fig. 145e-3). One of the best-studied systems involves the interaction of LPS from gram-negative bacteria and the GPI-anchored membrane protein CD14 found on the surface of professional phagocytes, including migrating and tissue-fixed macrophages and PMNs. A soluble form of CD14 is also found in plasma and on mucosal surfaces. A plasma protein, LPS-binding protein, transfers LPS to membrane-bound CD14 on myeloid cells and promotes binding of LPS to soluble CD14. Soluble CD14/LPS/LPS-binding protein complexes bind to many cell types and may be internalized to initiate cellular responses to microbial pathogens. It has been shown that peptidoglycan and lipoteichoic acid from gram-positive bacteria as well as cell-surface products of mycobacteria and spirochetes can interact with CD14 (Fig. 145e-3). Additional molecules, such as MD-2, also participate in the recognition of bacterial activators of inflammation.
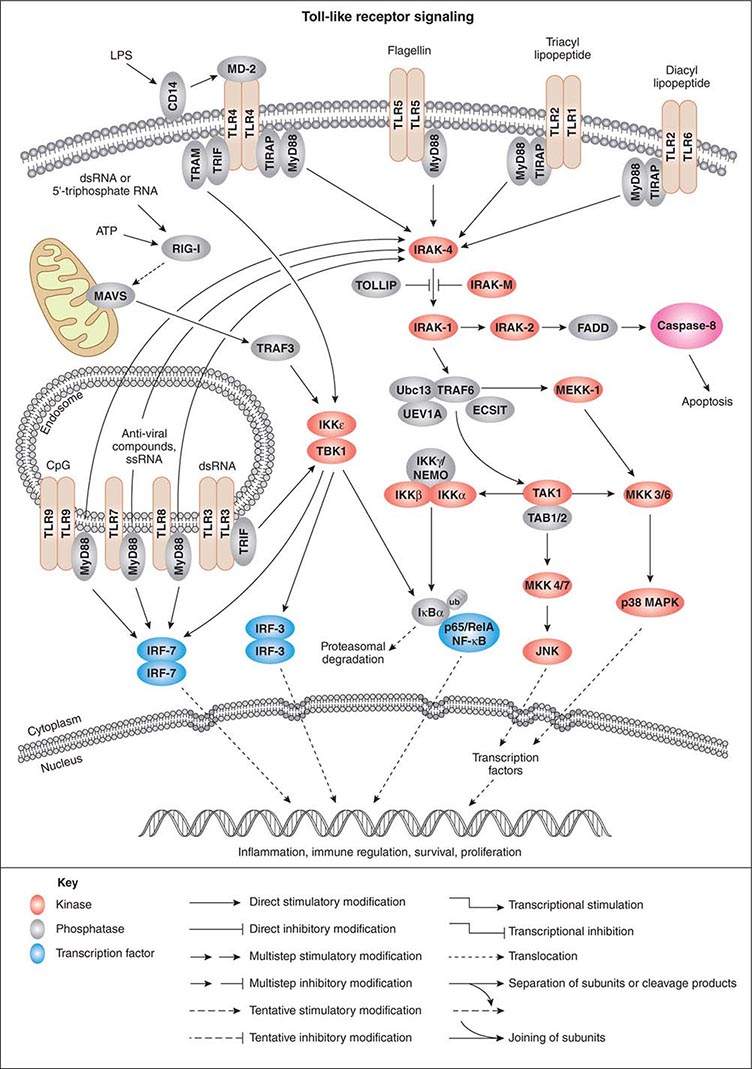
FIGURE 145e-3 Cellular signaling pathways for production of inflammatory cytokines in response to microbial products. Microbial cell-surface constituents interact with Toll-like receptors (TLRs), in some cases requiring additional factors such as MD-2, which facilitates the response to lipopolysaccharide (LPS) via TLR4. Although these constituents are depicted as interacting with the TLRs on the cell surface, TLRs contain extracellular leucine-rich domains that become localized to the lumen of the phagosome upon uptake of bacterial cells. The internalized TLRs can bind to microbial products. The TLRs are oligomerized, usually forming homodimers, and then bind to the general adapter protein MyD88 via the C-terminal Toll/IL-1R (TIR) domains, which also bind to TIRAP (TIR domain-containing adapter protein), a molecule that participates in the transduction of signals from TLRs 1, 2, 4, and 6. The MyD88/TIRAP complex activates signal-transducing molecules such as IRAK-4 (IL-1Rc-associated kinase 4), which in turn activates IRAK-1. This activation can be blocked by IRAK-M and Toll-interacting protein (TOLLIP). IRAK-1 activates TRAF6 (tumor necrosis factor receptor–associated factor 6), TAK1 (transforming growth factor β–activating kinase 1), and TAB1/2 (TAK1-binding protein 1/2). This signaling complex associates with the ubiquitin-conjugating enzyme Ubc13 and the Ubc-like protein UEV1A to catalyze the formation of a polyubiquitin chain on TRAF6. Polyubiquitination of TRAF6 activates TAK1, which, along with TAB1/2 (a protein that binds to lysine residue 63 in polyubiquitin chains via a conserved zinc-finger domain), phosphorylates the inducible kinase complex: IKKα, IKKβ, and IKKγ. IKKγ is also called NEMO (nuclear factor κB [NF-κB] essential modulator). This large complex phosphorylates the inhibitory component of NF-κB, IκBα, resulting in release of IκBα from NF-κB. Phosphorylated (PP) IκB is then ubiquitinated (ub) and degraded, and the two components of NF-κB, p50 or Rel and p65, translocate to the nucleus, where they bind to regulatory transcriptional sites on target genes, many of which encode inflammatory proteins. In addition to inducing NF-κB nuclear translocation, the TAK1/TAB1/2 complex activates MAP kinase transducers such as MKK 4/7 and MKK 3/6, which can lead to nuclear translocation of transcription factors such as AP1. TLR4 can also activate NF-κB nuclear translocation via the MyD88-independent TRIF (TIR domain–containing adapter-inducing IFN-β) and TRAM (TRIF-related adapter molecule) cofactors. Intracellular TLRs 3, 7, 8, and 9 also use MyD88 and TRIF to activate IFN response factors 3 and 7 (IRF-3 and IRF-7), which also function as transcriptional factors in the nucleus. ATP, adenosine 5’-triphosphate; ECSIT, evolutionarily conserved signaling intermediate in Toll pathways; FADD, Fas-associated protein with death domain; JNK, c-Jun N-terminal kinase; MAVS, mitochondrial antiviral signaling protein; MEKK-1, MAP/ERK kinase kinase 1; p38 MAPK, p38 mitogen-activated protein kinase; RIG-1, retinoic acid–inducible gene 1; TBK1, TANK-binding kinase 1. (Pathway diagram reproduced courtesy of Cell Signaling Technology, Inc. [www.cellsignal.com].)
GPI-anchored receptors do not have intracellular signaling domains; therefore, it is the TLRs that transduce signals for cellular activation due to LPS binding. Binding of microbial factors to TLRs to activate signal transduction occurs in the phagosome—and not on the surface—of dendritic cells that have internalized the microbe. This binding is probably due to the release of the microbial surface factor from the cell in the environment of the phagosome, where the liberated factor can bind to its cognate TLRs. TLRs initiate cellular activation through a series of signal-transducing molecules (Fig. 145e-3) that lead to nuclear translocation of the transcription factor NF-κB (nuclear factor κB), a master-switch for production of important inflammatory cytokines such as tumor necrosis factor α (TNF-α) and interleukin (IL) 1.
The initiation of inflammation can occur not only with LPS and peptidoglycan but also with viral particles and other microbial products such as polysaccharides, enzymes, and toxins. Bacterial flagella activate inflammation by binding of a conserved sequence to TLR5. Some pathogens (e.g., Campylobacter jejuni, Helicobacter pylori, and Bartonella bacilliformis) make flagella that lack this sequence and do not bind to TLR5; thus efficient host responses to infection are prevented. Bacteria also produce a high proportion of DNA molecules with unmethylated CpG residues that activate inflammation through TLR9. TLR3 recognizes double-stranded RNA, a pattern-recognition molecule produced by many viruses during their replicative cycle. TLR1 and TLR6 associate with TLR2 to promote recognition of acylated microbial proteins and peptides.
The myeloid differentiation factor 88 (MyD88) molecule and the Toll/IL-1R (TIR) domain-containing adapter protein (TIRAP) bind to the cytoplasmic domains of TLRs and also to receptors that are part of the IL-1 receptor families. Numerous studies have shown that MyD88/TIRAP-mediated transduction of signals from TLRs and other receptors is critical for innate resistance to infection, activating MAP-kinases and NF-κB and thereby leading to production of cytokines/chemokines. Mice lacking MyD88 are more susceptible than normal mice to infections with a broad range of pathogens. In one study, nine children homozygous for defective MyD88 genes had recurrent infections with S. pneumoniae, S. aureus, and P. aeruginosa—three bacterial species showing increased virulence in MyD88-deficient mice; however, unlike these mice, the MyD88-deficient children seemed to have no greater susceptibility to other bacteria, viruses, fungi, or parasites. Another component of the MyD88-dependent signaling pathway is a molecule known as IL-1 receptor–associated kinase 4 (IRAK-4). Individuals with a homozygous deficiency in genes encoding this protein are at increased risk for S. pneumoniae and S. aureus infections and, to some degree, for P. aeruginosa infections as well.
In addition to their role in MyD88-mediated signaling, some TLRs (e.g., TLR3 and TLR4) can activate signal transduction via a MyD88-independent pathway involving TIR domain–containing, adapter-inducing IFN-β (TRIF) and the TRIF-related adapter molecule (TRAM). Signaling through TRIF and TRAM activates the production of both NF-κB-dependent cytokines/chemokines and type 1 IFNs. The type 1 IFNs bind to the IFN-α receptor composed of two protein chains, IFNAR1 and IFNAR2. Humans produce three type 1 IFNs: IFN-α, IFN-β, and IFN-γ. These molecules activate another class of proteins known as the signal transducer and activator of transcription (STAT) complexes. The STAT factors are important in regulating immune system genes and thus play a critical role in responding to microbial infections.
Another intracellular complex of proteins found to be a major factor in the host cell response to infection is the inflammasome (Fig. 145e-4), in which inflammatory cytokines IL-1 and IL-18 are changed from their precursor to their active forms prior to secretion by the cysteine protease caspase-1. Within the inflammasome are additional proteins that are members of the nucleotide binding and oligomerization domain (NOD)–like receptor (NLR) family. Like the TLRs, NOD proteins sense the presence of the conserved microbial factors released inside a cell. Recognition of these PAMPs by NLRs leads to caspase-1 activation and to secretion of active IL-1 and IL-18 by an unknown mechanism. Studies of mice indicate that as many as four inflammasomes with different components are formed: the IPAF inflammasome, the NALP1 inflammasome, the cryopyrin/NALP3 inflammasome, and an inflammasome triggered by Francisella tularensis infection (Fig. 145e-4). The components depend on the type of stimulus driving inflammasome formation and activation.
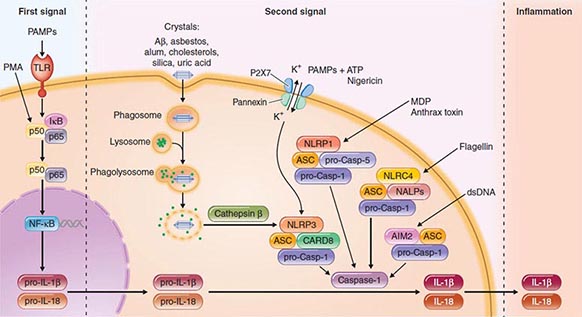
FIGURE 145e-4 Inflammasomes. The nucleotide-binding oligomerization domain-like receptor (NLR) family of proteins is involved in the regulation of innate immune responses. These proteins sense pathogen-associated molecular patterns (PAMPs) in the cytosol as well as the host-derived signals known as damage-associated molecular patterns (DAMPs). Certain NLRs induce the assembly of large caspase-1-activating complexes called inflammasomes. Activation of caspase-1 through autoproteolytic maturation leads to the processing and secretion of the proinflammatory cytokines interleukin 1β (IL-1β) and IL-18. So far, four inflammasomes have been identified and defined by the NLR protein that they contain: the NLRP1/NALP1b inflammasome; the NLRC4/IPAF inflammasome; the NLRP3/NALP3 inflammasome; and the AIM2 (absent in melanoma 2)–containing inflammasome. Aβ, amyloid β; ASC, apoptosis-associated speck-like protein containing CARD; ATP, adenosine 5’-triphosphate; CARD8, caspase recruitment domain–containing protein 8; IκB, inhibitor of κB; IPAF, interleukin-converting enzyme protease-activating factor; MDP, muramyl dipeptide; NF-κB, nuclear factor κB; P2X7, purinergic P2X7 (receptor); PMA, phorbol myristate acetate; TLR, Toll-like receptor. (Pathway diagram reproduced with permission from Invivogen [www.invivogen.com/review-inflammasome].)
A recent addition to the identified intracellular components responding to microbial infection is autophagy, initially described as an intracellular process for degradation and recycling of cellular components for reuse. Now it is clear that autophagy constitutes an early defense mechanism in which, after ingestion, microbial pathogens either within vacuoles or in the cytoplasm are delivered to lysosomal compartments for degradation. Avoidance of this process is critical if pathogens are to cause disease and can be achieved by multiple mechanisms, such as inhibition of proteins within the autophagic vacuole by shigellae, recruitment of host proteins to mask Listeria monocytogenes, and inhibition of formation of the vacuole by L. pneumophila.
ADDITIONAL INTERACTIONS OF MICROBIAL PATHOGENS AND PHAGOCYTES Other ways that microbial pathogens avoid destruction by phagocytes include production of factors that are toxic to these cells or that interfere with their chemotactic and ingestion function. Hemolysins, leukocidins, and the like are microbial proteins that can kill phagocytes that are attempting to ingest organisms elaborating these substances. For example, S. aureus elaborates a family of bicomponent leukocidins that bind to host receptors such as the HIV co-receptor CCR5 (which is also used by the LukE/D toxin) or—in the case of the Panton-Valentine leukocidin—the receptor of the C5a component of activated complement (which is used by LukF/S). Streptolysin O made by S. pyogenes binds to cholesterol in phagocyte membranes and initiates a process of internal degranulation, with the release of normally granule-sequestered toxic components into the phagocyte’s cytoplasm. E. histolytica, an intestinal protozoan that causes amebic dysentery, can disrupt phagocyte membranes after direct contact via the release of protozoal phospholipase A and pore-forming peptides.
MICROBIAL SURVIVAL INSIDE PHAGOCYTES Many important microbial pathogens use a variety of strategies to survive inside phagocytes (particularly macrophages) after ingestion. Inhibition of fusion of the phagocytic vacuole (the phagosome) containing the ingested microbe with the lysosomal granules containing antimicrobial substances (the lysosome) allows Mycobacterium tuberculosis, S. enterica serovar Typhi, and Toxoplasma gondii to survive inside macrophages. Some organisms, such as L. monocytogenes, escape into the phagocyte’s cytoplasm to grow and eventually spread to other cells. Resistance to killing within the macrophage and subsequent growth are critical to successful infection by herpes-type viruses, measles virus, poxviruses, Salmonella, Yersinia, Legionella, Mycobacterium, Trypanosoma, Nocardia, Histoplasma, Toxoplasma, and Rickettsia. Salmonella species use a master regulatory system—in which the PhoP/PhoQ genes control other genes—to enter and survive within cells, with intracellular survival entailing structural changes in the cell envelope LPS.
TISSUE INVASION AND TISSUE TROPISM
Tissue Invasion Most viral pathogens cause disease by growth at skin or mucosal entry sites, but some pathogens spread from the initial site to deeper tissues. Virus can spread via the nerves (rabies virus) or plasma (picornaviruses) or within migratory blood cells (poliovirus, Epstein-Barr virus, and many others). Specific viral genes determine where and how individual viral strains can spread.
Bacteria may invade deeper layers of mucosal tissue via intracellular uptake by epithelial cells, traversal of epithelial cell junctions, or penetration through denuded epithelial surfaces. Among virulent Shigella strains and invasive strains of E. coli, outer-membrane proteins are critical to epithelial cell invasion and bacterial multiplication. Neisseria and Haemophilus species penetrate mucosal cells by poorly understood mechanisms before dissemination into the bloodstream. Staphylococci and streptococci elaborate a variety of extracellular enzymes, such as hyaluronidase, lipases, nucleases, and hemolysins, that are probably important in breaking down cellular and matrix structures and allowing the bacteria access to deeper tissues and blood. For example, staphylococcal α-hemolysin binds to a receptor, A-disintegrin and metalloprotease 10 (ADAM-10), to cause endothelial cell damage and disruption of vascular barrier function—events that are likely critical for systemic spread of S. aureus from an initial infectious site. Organisms that colonize the gastrointestinal tract can often translocate through the mucosa into the blood and, under circumstances in which host defenses are inadequate, cause bacteremia. Yersinia enterocolitica can invade the mucosa through the activity of the invasin protein. The complex milieu of the basement membrane–containing structures, such as laminin and collagen, that anchor epithelial cells to mucosal surfaces must often be breached. Numerous organisms express factors known as MSCRAMMs (microbial surface components recognizing adhesive matrix molecules). These MSCRAMMS promote bacterial attachment to factors in the host extracellular matrix, such as laminin, collagen, and fibronectin. Additional microbial proteases, along with the host’s own surface-bound plasminogen and host matrix metalloproteases, then combine to degrade the extracellular matrix and promote microbial spread. Some bacteria (e.g., brucellae) can be carried from a mucosal site to a distant site by phagocytic cells that ingest but fail to kill the bacteria.
Fungal pathogens almost always take advantage of host immunocompromise to spread hematogenously to deeper tissues. The AIDS epidemic has resoundingly illustrated this principle: the immunodeficiency of many HIV-infected patients permits the development of life-threatening fungal infections of the lung, blood, and brain. Other than the capsule of C. neoformans, specific fungal antigens involved in tissue invasion are not well characterized. Both fungal pathogens and protozoal pathogens (e.g., Plasmodium species and E. histolytica) undergo morphologic changes to spread within a host. C. albicans undertakes a yeast-hyphal transformation wherein the hyphal forms are found where the fungus is infiltrating the mucosal barrier of tissues, while the yeast form grows on epithelial cell surfaces as well as on the tips of hyphae that have infiltrated tissues. Malarial parasites grow in liver cells as merozoites and are released into the blood to invade erythrocytes and become trophozoites. E. histolytica is found as both a cyst and a trophozoite in the intestinal lumen, through which this pathogen enters the host, but only the trophozoite form can spread systemically to cause amebic liver abscesses. Other protozoal pathogens, such as T. gondii, Giardia lamblia, and Cryptosporidium, also undergo extensive morphologic changes after initial infection to spread to other tissues.
Tissue Tropism The propensity of certain microbes to cause disease by infecting specific tissues has been known since the early days of bacteriology, yet the molecular basis for this propensity is understood somewhat better for viral pathogens than for other agents of infectious disease. Specific receptor-ligand interactions clearly underlie the ability of certain viruses to enter cells within tissues and disrupt normal tissue function, but the mere presence of a receptor for a virus on a target tissue is not sufficient for tissue tropism. Factors in the cell, route of viral entry, viral capacity to penetrate into cells, viral genetic elements that regulate gene expression, and pathways of viral spread in a tissue all affect tissue tropism. Some viral genes are best transcribed in specific target cells, such as hepatitis B genes in liver cells and Epstein-Barr virus genes in B lymphocytes. The route of inoculation of poliovirus determines its neurotropism, although the molecular basis for this circumstance is not understood.
Compared with viral tissue tropism, the tissue tropism of bacterial and parasitic infections has not been as clearly elucidated, but studies of Neisseria species have provided insights. Both N. gonorrhoeae, which colonizes and infects the human genital tract, and N. meningitidis, which principally colonizes the human oropharynx but can spread to the brain, produce type IV pili (Tfp) that mediate adherence to host tissues. In the case of N. gonorrhoeae, the Tfp bind to a glucosamine-galactose-containing adhesin on the surface of cervical and urethral cells; in the case of N. meningitidis, the Tfp bind to cells in the human meninges and thus cross the blood-brain barrier. N. meningitidis expresses a capsular polysaccharide, while N. gonorrhoeae does not; however, there is no indication that this property plays a role in the different tissue tropisms displayed by these two bacterial species. N. gonorrhoeae can use cytidine monophosphate N-acetylneuraminic acid from host tissues to add N-acetylneuraminic acid (sialic acid) to its lipooligosaccharide O side chain, and this alteration appears to make the organism resistant to host defenses. Lactate, present at high levels on genital mucosal surfaces, stimulates sialylation of gonococcal lipooligosaccharide. Bacteria with sialic acid sugars in their capsules, such as N. meningitidis, E. coli K1, and group B streptococci, have a propensity to cause meningitis, but this generalization has many exceptions. For example, all recognized serotypes of group B streptococci contain sialic acid in their capsules, but only one serotype (III) is responsible for most cases of group B streptococcal meningitis. Moreover, both H. influenzae and S. pneumoniae can readily cause meningitis, but these organisms do not have sialic acid in their capsules.
TISSUE DAMAGE AND DISEASE
Disease is a complex phenomenon resulting from tissue invasion and destruction, toxin elaboration, and host response. Viruses cause much of their damage by exerting a cytopathic effect on host cells and inhibiting host defenses. The growth of bacterial, fungal, and protozoal parasites in tissue, which may or may not be accompanied by toxin elaboration, can compromise tissue function and lead to disease. For some bacterial and possibly some fungal pathogens, toxin production is one of the best-characterized molecular mechanisms of pathogenesis, while host factors such as IL-1, TNF-α, kinins, inflammatory proteins, products of complement activation, and mediators derived from arachidonic acid metabolites (leukotrienes) and cellular degranulation (histamines) readily contribute to the severity of disease.
Viral Disease Viral pathogens are well known to inhibit host immune responses by a variety of mechanisms. Immune responses can be affected by decreasing production of most major histocompatibility complex molecules (adenovirus E3 protein), by diminishing cytotoxic T cell recognition of virus-infected cells (Epstein-Barr virus EBNA1 antigen and cytomegalovirus IE protein), by producing virus-encoded complement receptor proteins that protect infected cells from complement-mediated lysis (herpesvirus and vaccinia virus), by making proteins that interfere with the action of IFN (influenza virus and poxvirus), and by elaborating superantigen-like proteins (mouse mammary tumor virus and related retroviruses and the rabies nucleocapsid). Superantigens activate large populations of T cells that express particular subsets of the T cell receptor β protein, causing massive cytokine release and subsequent host reactions. Another molecular mechanism of viral virulence involves the production of peptide growth factors for host cells, which disrupt normal cellular growth, proliferation, and differentiation. In addition, viral factors can bind to and interfere with the function of host receptors for signaling molecules. Modulation of cytokine production during viral infection can stimulate viral growth inside cells with receptors for the cytokine, and virus-encoded cytokine homologues (e.g., the Epstein-Barr virus BCRF1 protein, which is highly homologous to the immunoinhibitory IL-10 molecule) can potentially prevent immune-mediated clearance of viral particles. Viruses can cause disease in neural cells by interfering with levels of neurotransmitters without necessarily destroying the cells, or they may induce either programmed cell death (apoptosis) to destroy tissues or inhibitors of apoptosis to allow prolonged viral infection of cells. For infection to spread, many viruses must be released from cells. In a newly identified function, viral protein U (Vpu) of HIV facilitates the release of virus, a process that is specific to certain cells. Mammalian cells produce a restriction factor involved in inhibiting the release of virus; for HIV, this factor is designated BST-2 (bone marrow stromal antigen 2)/HM1.24/CD317, or tetherin. Vpu of HIV interacts with tetherin, promoting release of infectious virus. Overall, disruption of normal cellular and tissue function due to viral infection, replication, and release promotes clinical disease.
Bacterial Toxins Among the first infectious diseases to be understood were those due to toxin-elaborating bacteria. Diphtheria, botulism, and tetanus toxins are responsible for the diseases associated with local infections due to Corynebacterium diphtheriae, Clostridium botulinum, and Clostridium tetani, respectively. Clostridium difficile is an anaerobic gram-positive organism that elaborates two toxins, A and B, responsible for disruption of the intestinal mucosa when organism numbers expand in the intestine, leading to antibiotic-associated diarrhea and potentially to pseudomembranous colitis. Enterotoxins produced by E. coli, Salmonella, Shigella, Staphylococcus, and V. cholerae contribute to diarrheal disease caused by these organisms. Staphylococci, streptococci, P. aeruginosa, and Bordetella elaborate various toxins that cause or contribute to disease, including toxic shock syndrome toxin 1; erythrogenic toxin; exotoxins A, S, T, and U; and pertussis toxin. A number of bacterial toxins (e.g., cholera toxin, diphtheria toxin, pertussis toxin, E. coli heat-labile toxin, and P. aeruginosa exotoxin) have adenosine diphosphate ribosyl transferase activity; i.e., the toxins enzymatically catalyze the transfer of the adenosine diphosphate ribosyl portion of nicotinamide adenine diphosphate to target proteins and inactivate them. The staphylococcal enterotoxins, toxic shock syndrome toxin 1, and the streptococcal pyogenic exotoxins behave as superantigens, stimulating certain T cells to proliferate without processing of the protein toxin by antigen-presenting cells. Part of this process involves stimulation of the antigen-presenting cells to produce IL-1 and TNF-α, which have been implicated in many clinical features of diseases like toxic shock syndrome and scarlet fever. A number of gram-negative pathogens (Salmonella, Yersinia, and P. aeruginosa) can inject toxins directly into host target cells by means of a complex set of proteins referred to as the type III secretion system. Loss or inactivation of this virulence system usually greatly reduces the capacity of a bacterial pathogen to cause disease.
Endotoxin The lipid A portion of gram-negative LPS has potent biologic activities that cause many of the clinical manifestations of gram-negative bacterial sepsis, including fever, muscle proteolysis, uncontrolled intravascular coagulation, and shock. The effects of lipid A appear to be mediated by the production of potent cytokines due to LPS binding to CD14 and signal transduction via TLRs, particularly TLR4. Cytokines exhibit potent hypothermic activity through effects on the hypothalamus; they also increase vascular permeability, alter the activity of endothelial cells, and induce endothelial-cell procoagulant activity. Numerous therapeutic strategies aimed at neutralizing the effects of endotoxin are under investigation, but so far the results have been disappointing. It has been suggested that this lack of success may be due to substantial differences between mouse and human inflammatory responses to factors such as endotoxin; thus drugs developed in mouse models of infection may not be applicable to the human response.
Invasion Many diseases are caused primarily by pathogens growing in tissue sites that are normally sterile. Pneumococcal pneumonia is mostly attributable to the growth of S. pneumoniae in the lung and the attendant host inflammatory response, although specific factors that enhance this process (e.g., pneumolysin) may be responsible for some of the pathogenic potential of the pneumococcus. Disease that follows bloodstream infection and invasion of the meninges by meningitis-producing bacteria such as N. meningitidis, H. influenzae, E. coli K1, and group B streptococci appears to be due solely to the ability of these organisms to gain access to these tissues, multiply in them, and provoke cytokine production leading to tissue-damaging host inflammation.
Specific molecular mechanisms accounting for tissue invasion by fungal and protozoal pathogens are less well described. Except for studies pointing to factors like capsule and melanin production by C. neoformans and possibly levels of cell wall glucans in some pathogenic fungi, the molecular basis for fungal invasiveness is not well defined. Melanism has been shown to protect the fungal cell against death caused by phagocyte factors such as nitric oxide, superoxide, and hypochlorite. Morphogenic variation and production of proteases (e.g., the Candida aspartyl proteinase) have been implicated in fungal invasion of host tissues.
If pathogens are to effectively invade host tissues (particularly the blood), they must avoid the major host defenses represented by complement and phagocytic cells. Bacteria most often elude these defenses through their surface polysaccharides—either capsular polysaccharides or long O-side-chain antigens characteristic of the smooth LPS of gram-negative bacteria. These molecules can prevent the activation and/or deposition of complement opsonins or can limit the access of phagocytic cells with receptors for complement opsonins to these molecules when they are deposited on the bacterial surface below the capsular layer. Another potential mechanism of microbial virulence is the ability of some organisms to present the capsule as an apparent self antigen through molecular mimicry. For example, the polysialic acid capsule of group B N. meningitidis is chemically identical to an oligosaccharide found on human brain cells.
Immunochemical studies of capsular polysaccharides have led to an appreciation of the tremendous chemical diversity that can result from the linking of a few monosaccharides. For example, three hexoses can link up in more than 300 different, potentially serologically distinct ways, while three amino acids have only six possible peptide combinations. Capsular polysaccharides have been used as effective vaccines against meningococcal meningitis as well as against pneumococcal and H. influenzae infections and may prove to be of value as vaccines against any organisms that express a nontoxic, immunogenic capsular polysaccharide. In addition, most encapsulated pathogens become virtually avirulent when capsule production is interrupted by genetic manipulation; this observation emphasizes the importance of this structure in pathogenesis. It is noteworthy that the capsule-like surface polysaccharide PNAG has been found as a conserved structure shared by many microbes but generally is a poor target for antibody-mediated immunity because of the propensity of most humans and animals—all colonized by PNAG-producing microbes—to produce a nonprotective type of antibody. Altering the structure of PNAG by removing the acetate substituents on the N-acetylglucosamine monomers yields an immunogenic form, deacetylated PNAG, that reportedly induces antibodies that protect animals against diverse microbial pathogens.
Host Response The inflammatory response of the host is critical for interruption and resolution of the infectious process but is often responsible for the signs and symptoms of disease. Infection promotes a complex series of host responses involving the complement, kinin, and coagulation pathways. The production of cytokines such as IL-1, IL-18, TNF-α, IFN-γ, and other factors regulated in part by the NF-κB transcription factor leads to fever, muscle proteolysis, and other effects. An inability to kill or contain the microbe usually results in further damage due to the progression of inflammation and infection. For example, in many chronic infections, degranulation of host inflammatory cells can lead to release of host proteases, elastases, histamines, and other toxic substances that can degrade host tissues. Chronic inflammation in any tissue can lead to the destruction of that tissue and to clinical disease associated with loss of organ function, such as sterility from pelvic inflammatory disease caused by chronic infection with N. gonorrhoeae.
The nature of the host response elicited by the pathogen often determines the pathology of a particular infection. Local inflammation produces local tissue damage, while systemic inflammation, such as that seen during sepsis, can result in the signs and symptoms of septic shock. The severity of septic shock is associated with the degree of production of host effectors. Disease due to intracellular parasitism results from the formation of granulomas, wherein the host attempts to wall off the parasite inside a fibrotic lesion surrounded by fused epithelial cells that make up so-called multinucleated giant cells. A number of pathogens, particularly anaerobic bacteria, staphylococci, and streptococci, provoke the formation of an abscess, probably because of the presence of zwitterionic surface polysaccharides such as the capsular polysaccharide of Bacteroides fragilis. The outcome of an infection depends on the balance between an effective host response that eliminates a pathogen and an excessive inflammatory response that is associated with an inability to eliminate a pathogen and with the resultant tissue damage that leads to disease.
TRANSMISSION TO NEW HOSTS
As part of the pathogenic process, most microbes are shed from the host, often in a form infectious for susceptible individuals. However, the rate of transmissibility may not necessarily be high, even if the disease is severe in the infected individual, as these traits are not linked. Most pathogens exit via the same route by which they entered: respiratory pathogens by aerosols from sneezing or coughing or through salivary spread, gastrointestinal pathogens by fecal-oral spread, sexually transmitted diseases by venereal spread, and vector-borne organisms by either direct contact with the vector through a blood meal or indirect contact with organisms shed into environmental sources such as water. Microbial factors that specifically promote transmission are not well characterized. Respiratory shedding is facilitated by overproduction of mucous secretions, with consequently enhanced sneezing and coughing. Diarrheal toxins such as cholera toxin, E. coli heat-labile toxins, and Shigella toxins probably facilitate fecal-oral spread of microbial cells in the high volumes of diarrheal fluid produced during infection. The ability to produce phenotypic variants that resist hostile environmental factors (e.g., the highly resistant cysts of E. histolytica shed in feces) represents another mechanism of pathogenesis relevant to transmission. Blood parasites such as Plasmodium species change phenotype after ingestion by a mosquito—a prerequisite for the continued transmission of this pathogen. Venereally transmitted pathogens may undergo phenotypic variation due to the production of specific factors to facilitate transmission, but shedding of these pathogens into the environment does not result in the formation of infectious foci.
SUMMARY
In summary, the molecular mechanisms used by pathogens to colonize, invade, infect, and disrupt the host are numerous and diverse. Each phase of the infectious process involves a variety of microbial and host factors interacting in a manner that can result in disease. Recognition of the coordinated genetic regulation of virulence factor elaboration when organisms move from their natural environment into the mammalian host emphasizes the complex nature of the host-parasite interaction. Fortunately, the need for diverse factors in successful infection and disease implies that a variety of therapeutic strategies may be developed to interrupt this process and thereby to prevent and treat microbial infections.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


