The measurement of antibiotic concentrations in various fluids has been a prominent aspect of the evaluation of new antibiotics and the quality control of their manufacture. With the availability of rapid, accurate assays, the measurement of antibiotic material in serum and other body fluids is feasible, desirable, and widely practiced for these purposes. Clinically, such assays have been used primarily for the determination of aminoglycoside levels in serum, for which peak levels primarily establish adequacy of therapy, while trough levels reflect potential drug accumulation and toxicity.
Although the most common reason for performing rapid serum antibiotic assays in hospitals is to regulate therapy with aminoglycoside antibiotics and vancomycin, there are other indications. For patients who have organ dysfunction, such as hepatic or renal failure, it is desirable to know whether or not antibiotics that are metabolized or eliminated (e.g., CAM) by damaged or imperfectly functioning organs accumulate. In some patients who are taking oral antibiotics, it may be important to know the extent to which they are absorbing the drugs from their gastrointestinal tracts (13–16). Therapeutic monitoring of β-lactam, macrolide, tetracycline, and other antibiotics with wide therapeutic windows occurs infrequently, but may be performed to assess compliance, and/or in patients with renal failure.
Aminoglycoside levels are usually ordered to establish the adequacy of therapy and to prevent toxicity. Retrospectively, there is a correlation between both peak and trough levels and toxicity. In one study, Black et al. (17) found a significant correlation between ototoxicity of amikacin and both peak and trough levels, with the P value being slightly lower for the peak than the trough value.
Another indication for the measurement of antibiotics from human body fluids has been the establishment of therapeutic levels of drugs in various body fluids. The field of the pharmacokinetics of antibiotics has developed rapidly, along with the number of drugs that are currently available to treat serious infections. The concept of the “class” or “type” antibiotic that is representative of all related members is no longer valid. For example, we can no longer predict the tissue distribution of all cephalosporins based on the activity of cephalothin. Many of the third-generation cephalosporins appear in extravascular body fluid compartments, while cephalothin does not. Therefore, the ability to assay each cephalosporin level in cerebrospinal fluid is desirable. As pharmaceutical chemists modify currently available antibiotics, additional generations and individual drugs will continue to be developed.
In the 1960s and 1970s, most work concentrated on microbiologic assay systems for the major classes of antibiotics. The 1970s witnessed the evolution of reference methods to rapid procedures that became available in clinical laboratories. By the late 1970s, approximately 75% to 85% of all assays in clinical laboratories were microbiologic in nature, with most of the remainder being RIAs (18). Subsequently, commercially available, nonisotopic immunoassays largely replaced microbiologic assays. Methods for the assay of virtually all classes of antibiotics by HPLC have become available and can be performed in a broad spectrum of laboratories. Immunoassays for aminoglycosides and vancomycin have become routine “black box” procedures because of the widespread availability of commercial kits. Assays for antituberculous, antifungal, and antiviral medications can be found in their respective chapters.
HOW TO CHOOSE AN ASSAY
The selection of an assay method depends on the clinical and research needs and on the capabilities and resources of the laboratory in which the assay is to be performed. For high-volume assays that require rapid turnaround, commercially sold immunoassays that use specific monoclonal antibodies and are performed on automated chemistry analyzers will be the methods of choice. Low-volume assays for which monoclonal antibodies, commercial kits, or automated chemistry analyzers are unavailable may be performed by HPLC or, in some instances, bioassay. As a rule of thumb, a laboratory that processes 10 or more specimens per day should select the most automated, least labor-intensive methods available, even if reagent and equipment costs are increased by doing so. Because the availability of assays to detect minimal deviations from the therapeutic range can be clinically important, sensitivity may take precedence over specificity (19). There remain circumstances in which microbiologic assays may still be useful with particular antibiotics. They have the advantage of being performed without costly or dedicated laboratory equipment. In addition, they determine the concentration of all active metabolites in one step.
There is an extremely broad array of microbiologic agar diffusion assays that are available for use. Each combination of agar, pH, and organism has been chosen to optimize the measurement of a given antibiotic, either alone or in combination with other antibiotics. For clinical purposes, however, the absolute sensitivity of the organism used is of less importance than such factors as the turnaround time of the test, the ability to store plates, and the resistance pattern of the organism. Rarely is it necessary for clinical assays to equal the sensitivity of research assays (20,21). When microbiologic assays in agar are well designed and measurements of zone sizes are made properly, these assays have shown excellent agreement with immunologic techniques (22–25).
A study was undertaken in Great Britain in the mid-1970s in which salted serum specimens of gentamicin were sent to numerous clinical laboratories that performed microbiologic assays for measurement of the drug in body fluids (26). This study classified the performance of fewer than 20% of laboratories, on average, as “good.” Those laboratories with the least experience fared the worst. Radioenzyme assays and RIAs were studied according to similar protocols. Repeated testing of the same laboratories demonstrated marked improvement because of either increased motivation or reexamination of and improvement in their techniques. Pocket calculators can be programmed to provide linear regression analysis with such factors as slope, intercept, and coefficient of variation, and it is recommended that such records be maintained as an internal quality control check of a bioassay (27). Deviations beyond 2 standard deviations (SDs), or 1 SD in the same direction, an inordinate number of times should prompt reexamination of the method.
Proficiency testing samples with serum samples spiked with antibiotics such as amikacin, gentamicin, tobramycin, and vancomycin are available from the College of American Pathologists and other authorized organizations. Participation in Centers for Medicare and Medicaid Services–approved proficiency testing programs for clinically tested antibiotic analytes for which such programs are available is required under the Clinical Laboratory Improvement Amendments of 1988 and for laboratory accreditation by certifying entities, irrespective of the testing method used. For antibiotics for which no Centers for Medicare and Medicaid Services–approved proficiency testing samples are available, laboratories must document biannual verification of the accuracy of their procedures.a
Table 8.2 summarizes the general advantages and disadvantages of the agar diffusion assay method. Almost all of the equipment necessary to perform the microbiologic assay successfully is familiar to microbiology technicians and is readily available in the microbiology laboratory. The only factor that needs to be rigidly controlled is the preparation of the antibiotic standards (28). For those laboratories without sensitive electrical balances or a person experienced in preparing such standards, the hospital pharmacy can be extremely helpful and is often willing to prepare standards. Skilled technical personnel are not required to perform the assay, and it generally takes less than 2 hours to train a technician to successfully complete it. Once the standards are made and familiarity with the calculation of results and preparation of media is attained, an antibiotic blood level can be set up within 10 to 20 minutes of receipt of the specimen. With most Staphylococcus, Enterobacteriaceae, and Bacillus assays, results are available within 2 to 4 hours (29–31). Finally, inexpensive equipment is used, limiting the cost of the assay and possible problems with instrument accessibility.
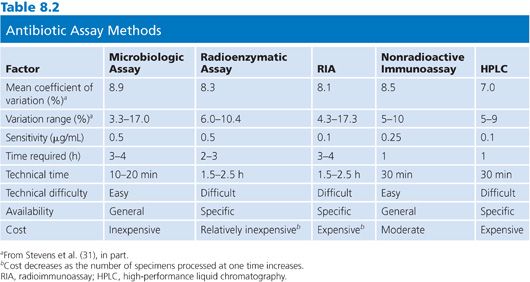
The major disadvantage of the microbiologic assay is the steep slope that is generated, especially for the assay of aminoglycoside antibiotics. As a consequence, a small difference in measurement can significantly alter the apparent concentration (32). Vernier calipers or automated instruments that are designed especially for this purpose, rather than millimeter rulers, should always be used to measure zone sizes. Rather than hand drawing a “best” straight line, x and y values should be interpolated from a regression curve. The regression curve can be generated with a pocket calculator. A second or third antibiotic that is present in the specimen and unknown to the laboratory can, of course, lead to erroneously high results. Because a large percentage of hospitalized patients receive two or more antibiotics, it is incumbent on the assayist to contact the ward or the pharmacy to inquire as to what antibiotics the patient is receiving, irrespective of the information that is provided on the requisition form (33,34).
It is probably convenient when choosing a microbiologic assay to choose an organism, such as Klebsiella, that is inherently resistant to many antibiotics. The technician should be sure that likely combinations of antibiotics will not act synergistically on the organism that is selected. Inordinately high results should be confirmed by repeating the assay with the specimen diluted 1:5 and 1:10 in normal human serum and by determining whether an unknown second antibiotic is present (35).
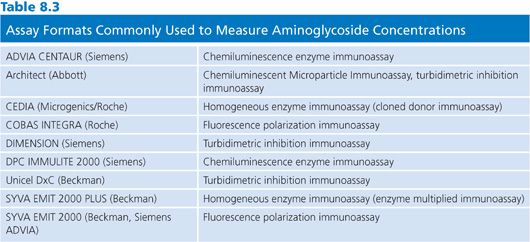
Immunoassays are the method of choice for aminocyclitol/aminoglycoside antibiotics. The major advantages of these assays include specificity, automated analysis, microprocessor-controlled calculations, and versatility of analytes that can be measured with the required instrumentation. Immunoassays for aminoglycosides and other antibiotics are invariably competitive assays, in which bound or conjugated drug competes with free drug in the specimen for antibody binding sites. Some assay formats that are commonly used in clinical laboratories to measure aminoglycoside concentrations are listed in Table 8.3. For the measurement of gentamicin and tobramycin concentrations, the Abbott AXSYM FPIA (Abbott Laboratories, Abbott Park, IL), the Dade Dimension turbidimetric inhibition immunoassay (Siemens Diagnostics, Tarrytown, NY), and the Beckman Synchron (Beckman Coulter, Inc, Brea, CA) reagent are among the most commonly used assay systems in clinical laboratories. For the analysis of amikacin concentrations, the Abbott TDX/TDX FLEX (Abbott Laboratories, Abbott Park, IL) is a very popular assay format.
Although the material costs per test are higher for immunoassays than for microbiologic assays, the labor costs are significantly less. The necessary instrumentation costs significantly less than RIA or radioenzymatic assay equipment, and, in most cases, capital expenditures for instruments can be transferred to the costs of individual tests as disposable items. Because many reagents have refrigerated lifetimes of up to 12 weeks after they are constituted, these methods are applicable to laboratories of any size. Moreover, specialized technologists are not needed to perform immunoassays. In deciding between various assay methods and formats, one often must balance the costs of labor versus the costs of supplies. For clinical purposes, the sensitivity of nonisotopic immunoassays is no different from that of RIAs (36–43).
HPLC has become the method of choice for analysis of β-lactam concentrations in serum and other body fluids. Again, one must deal with the proviso that the equipment is expensive. However, one can process large numbers of specimens quite accurately in short periods of time (44,45). HPLC technology is the primary method of analysis of antibiotic concentrations in human body fluids in the research setting, especially when new drugs are being investigated. It is the method of choice when one wishes to analyze individual components of an antibiotic or its metabolites (46).
A particular advantage of HPLC is the ability to quantify closely related compounds in a mixture (47–53). HPLC procedures have been developed for the analysis of almost all antibiotics used for the treatment of human diseases (54–56). Unfortunately, the high cost of HPLC instruments makes it impractical for this technique to compete with immunoassays once a specific antibody has been produced for an antibiotic. In addition to the cost of the HPLC apparatus, the procedure generally requires a trained technologist and dedicated instrument to the analysis of particular antibiotics or classes of antibiotics for a defined period of time. Different antibiotic classes may require different columns and conditions. Unlike gas-liquid chromatography (GLC), however, the basic columns in HPLC technology (C8 and C18) may be used in the reverse-phase mode for all water-soluble antibiotics. The primary modifications that are required for the measurement of these drug concentrations are to the solvent systems. Because of the ability of HPLC to separate constituents in a mixture (e.g., the three gentamicin components), its analytical capabilities extend beyond those of RIAs (57–60).
SPECIMEN HANDLING
To achieve optimal results, specimens should be processed as soon as possible after they are obtained. Each antibiotic loses potency at its own rate. For example, aminoglycosides are much more stable than the penicillins.
Serum or ethylenediaminetetraacetic acid (EDTA)–treated plasma is the recommended specimen for measuring blood concentrations of aminoglycosides. Heparin in the concentrations present in heparin-containing blood collection tubes may inactivate aminoglycoside antibiotics through complex formation. This can cause underestimates of aminoglycoside concentrations. Heparin in therapeutic concentrations does not appear to have in vivo effects on aminoglycoside activity, but heparin-containing collection tubes should not be used unless their suitability has been verified by the manufacturer of an assay. Although sera of patients receiving gentamicin can be stored between 20°C and +25°C for up to 2 days without any significant effect, samples that are not tested within 2 hours should be stored at 0°C to 5°C to avoid possible inactivation by coadministered β-lactam antibiotics (61,62).
Acceptable samples for measurement of blood CAM concentrations include serum and EDTA or citrate anticoagulated plasma. These specimens should be kept protected from light and, if not analyzed immediately, stored frozen.
Serum or EDTA anticoagulated plasma are appropriate specimens for analysis of vancomycin concentrations. Heparin-containing samples are generally also considered acceptable. However, reports of vancomycin instability in the presence of heparin recommend exercising caution in testing such samples. Verification of specimen suitability is essential.
In general, if a specimen cannot be processed within 1 to 2 hours of its receipt, steps must be taken to ensure its potency. If it is to be processed the same day, it should be refrigerated at less than 4°C. If it cannot be processed the same day, it should be frozen. If the specimen can be processed within 3 to 4 days, freezing at −20°C should be sufficient. If storage is for a longer period, the specimen should be frozen at −70°C. If a tissue specimen cannot be processed within 2 to 3 hours, it should be frozen at −20°C for processing within 24 hours. If storage will be for more than 24 hours, the specimen should be frozen at −70°C (63).
FLUIDS OTHER THAN BLOOD
The clinical assay of antibiotics from sites other than serum or plasma has generally not been standardized (64). The determination of antibiotic concentrations in fluids other than blood involves all the variability that accompanies the measurement of antibiotic levels in serum and much more (65–68). These fluids can contain different types and amounts of proteins, chemical compounds, and cellular compounds and have different pH values as compared with serum. In order for an assay to be valid, standards must be prepared in the same milieu as the sample or in a matrix that has been demonstrated to be equivalent to it. In assaying a fluid, one must be sure that the specimen is not contaminated by blood. Although rather simplistic in practice, the avoidance of such contamination is difficult and sometimes not readily detectable (69).
No universal method is described that can be used for the assay of antibiotic concentrations in all fluids. The key to the assay of antibiotics in body fluids is that the standard antibiotic dilution curves should be prepared in the same media as the patient samples. For example, in assaying the level of an antibiotic in joint fluid, one should prepare standards in normal human joint fluid or in a solvent with a high protein content. In the determination of gentamicin concentrations in spinal fluid, it was found that if the standards were made in water, a 400% error could result. If the standards were made in 0.5% saline, the error was reduced to 50%. Finally, if the suspending medium was 150 mmol/L NaCl per 4.5 mmol/L CaCl2, the results were not significantly different from those for cerebrospinal fluid (70).
One may determine the antibiotic concentration in fluids such as cerebrospinal fluid, joint fluid, or any other nonviscous fluid by diluting the antibiotic standards in normal fluid and performing the microbiologic assay, as described in the section “Elements Influencing Microbiologic Assays,” for total biologic activity (71). When assaying an antibiotic that is not readily degraded (e.g., aminoglycosides and CAM), an immunologic or chemical assay is best.
It is important to establish the concentration of antibiotic that was actually present in the fluid under study from that obtained in the presence of contaminating blood. One should analyze the fluid specimen for blood by weighing the fluid and measuring the amount of blood inside it (72). This may be done by spectrophotometric analysis. The fluid should be centrifuged at 3,000 rpm for 15 minutes and the supernatant placed in a 1-cm spectrophotometric cell. After brief aeration, the absorbance at 576 nm is recorded and corrected for turbidity produced by cellular debris by recording the absorbances at 600 and 624 nm. At these latter wavelengths, the hemoglobin absorption is less than 10% of the maximum.
To the absorbance at 600 nm is added the difference between the absorbances at 600 and 624 nm. The resulting absorbance represents the contributions made by the turbidity to the absorbance at 576 nm. This correction is then subtracted from total absorbance at 576 nm, with the remainder providing a measure of the tissue hemoglobin content. The absorbance value so obtained can be standardized in terms of hemoglobin concentration by making similar measurements with a sample of the patient’s own blood, diluted 1:150.
Another potential problem in the assay of antibiotic concentrations in tissue is storage. When performing fluid assays, one tends to collect the fluid and freeze it until ready for use. In contrast, most clinical assays of antibiotic blood levels are performed soon after the specimen has been received.
The stability of an antibiotic depends on the medium in which it is suspended. An antibiotic that is stable in buffer for long periods of time at 20°C or at 4°C may be quite unstable at the same temperature in serum or tissue (73). One should store all body fluids at −70°C prior to assay. It is incumbent on the assayist, when storing antibiotics in a given tissue medium, to store a standard in parallel to assess any loss of activity (74). At this time, it is not possible to definitively predict how an antibiotic’s stability will be affected by the particular fluid or tissue in which it is found (73,74). Antibiotics have been assayed successfully from many body sites by employing the principles described. Each fluid and antibiotic must be treated as a unique combination. Procedures that are satisfactory for one fluid and antibiotic pair will not necessarily prove satisfactory for another pair, even if the two are closely related.
Urine should be buffered to the optimum pH of the antibiotic in question, and its protein and sugar content should be recorded. Normal pooled urine from patients who are not receiving antibiotics, buffered in the same way, should be used as controls. The Bacillus subtilis technique has been used successfully for the determination of antibiotic concentrations in urine. As described in the section “Elements Influencing Microbiologic Assays,” dilutions in a phosphate buffer provide good results. It has been reported that individuals may naturally excrete organic acids that can be antibacterial. Although this scenario is not common, the assayist should be on guard for spuriously high urine antibiotic levels due to the presence of such organic acids. One must filter-sterilize the urine if it contains microorganisms (75,76).
MICROBIOLOGIC ASSAYS
Microbiologic assays are relative rather than absolute (77). In one type of assay, an antibiotic concentration is determined from the microbiologic response of a strain of test organism to a series of standard antibiotic concentrations.
Agar Methods
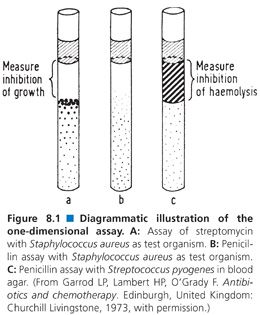
Assays in agar fall into three broad categories: one-dimensional, two-dimensional, and three-dimensional. The one-dimensional assay employs a test tube or a capillary tube in which seeded agar has been poured and allowed to harden as the agar medium (Fig. 8.1). An antibiotic test suspension is pipetted onto the surface of the hardened agar, and zones of inhibition in one dimension are formed. The one-dimensional assay is especially appropriate for the assay of antibiotics under anaerobic conditions. This method never gained popularity in clinical laboratories in the United States, although it has been used frequently in Japan (78). The method does not lend itself to automation. Its major disadvantages are that it requires complex sample preparation and time-consuming dilution steps. The one-dimensional assay can be useful in determining the antibiotic fluid levels in a pediatric population or in other circumstances in which only a small amount of fluid is available for testing. A procedure for the one-dimensional tube assay is provided later in this chapter.
Clinical laboratories have most commonly used two-dimensional or three-dimensional assays. A two-dimensional assay is one in which the antibiotic diffuses directly against a wall of seeded bacteria. This typically involves one of two designs. In the first technique, wells are cut in agar and seeded throughout with the test organism. In the second method, the test organism is swabbed onto the surface of the agar. Antibiotic disks are then placed on the agar surface. The assay is considered two-dimensional because the concentration of antibiotic as it diffuses radially is equal at any given distance from its source. In a three-dimensional assay, the antibiotic diffuses vertically to the bottom of the Petri plate, in addition to migrating along the surface of the agar. At a distance x from the disk, therefore, the concentration of antibiotic may not be identical in all dimensions. As the agar thickness in a three-dimensional system decreases, the likelihood that the antibiotic concentrations are equal at any given distance x from the source increases because the three-dimensional system is physically moving toward a two-dimensional system.
For clinical assays, the differences between the three- and two-dimensional systems are of little significance. In research settings, both systems should be compared to establish that they yield equal results before a three-dimensional system is used. The two-dimensional assay is theoretically somewhat sounder than the three-dimensional assay (79). The three-dimensional assay is one in which cylinders, fish spines, or disks are placed on the surfaces of seeded agar plates. The well-type, two-dimensional assay and paper disk-type, three-dimensional assay have been extensively used clinically. One generally can determine the lower limits of antibiotic concentrations using well-type assays.
Elements Influencing Microbiologic Assays
In performing microbiologic assays, one must carefully account for the many conditions that affect the action of the antibiotic under study and the growth properties of the organism. Deviations from the use of rigid controls result in erroneous assay values (80). The most prominent factors that affect the performance of microbiologic assays follow.
Design
Basic to the assay of antibiotic concentrations is a system design that determines the levels accurately. One must be certain that the zone of inhibition around a source of antibiotic is produced in direct proportion to the amount of antibiotic that is contained within that source. One must also be certain that the response is linear within the range that is normally encountered in clinical specimens. Because dose-response curves are sigmoidal in shape, the assayist must test a sufficient number of specimens to be assured that they fall on the dose-response curve. Environmental factors such as temperature and pH must not affect the assay in either the high or low ranges.
Media
There are a wide variety of media from which to choose for the assay of antibiotic concentrations. Grove and Randall (81) in 1955 described 11 different media that were available for the assay of antibiotic concentrations in human specimens. Almost all subsequently published methods have used some variation of these 11 types of media. The choice of medium is related to the antibiotic under study, the assay design, and the test organism. Although a detailed discussion of the relative merits of each medium–antibiotic–organism combination is not within the scope of this chapter, the medium chosen should be optimized for the assayed antibiotic (79) (Table 8.4). A medium that is satisfactory for use with one antibiotic–organism combination may not be acceptable for the measurement of the same antibiotic concentration in an assay that uses a different organism (82).
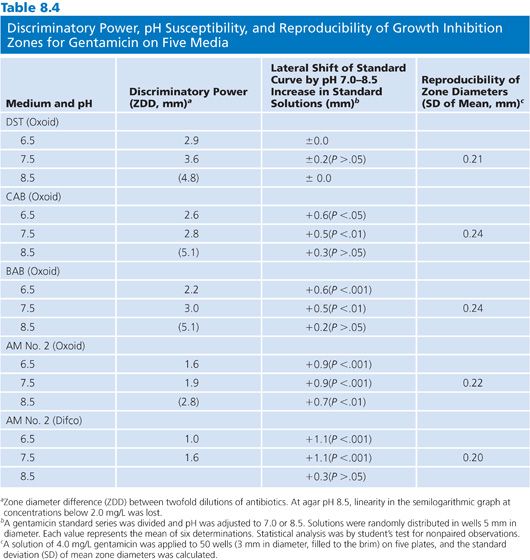
The pH of the agar may vary with the mixture of ingredients and the method of preparation. Although the agars are made according to the same formulation, they may not be identical. This variability is the result of the undefined nature of many of the ingredients. For example, yeast extract and animal infusions are in no way standardized. Table 8.4 presents the effects that different agar preparations and pH values have on the results of an assay for the measurement of gentamicin concentrations. Although most researchers have not thought it necessary to use indicators in media, several have found it useful to add fermentable sugars that enable the determination of zones of inhibition by the accompanying pH changes (83). The spraying of plates with tetrazolium blue allows for the ascertainment of zone size more rapidly than by visible inspection alone (84). Agars differ in their susceptibilities to pH changes, crispness of zones, absolute sensitivities, and slopes of the standard curves that they produce (85). Because antibiotic standards and patients’ specimens are included on the same plates in most clinical assays, such variations are internally controlled (Fig. 8.2).
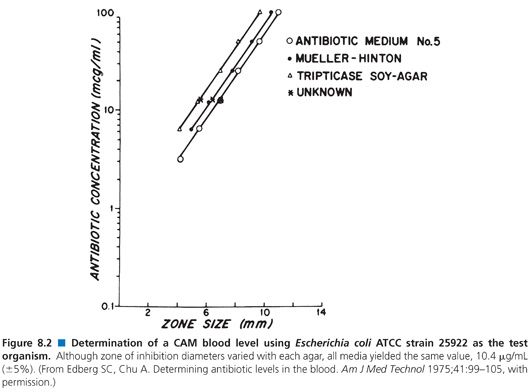
Antibiotic Standards
The production of antibiotic standards is the most critical factor in these assays. Because microbiologic assays are relative assays, any inconsistency in the antibiotic standards results in erroneous concentration measurements. Each antibiotic is differentially active at different pH values (78,86). Because it is impractical to make antibiotic standards from powder each time an assay is to be performed, one may produce and store working concentrations of the antibiotic in small aliquots. Practically, the antibiotics are dissolved in appropriate buffers (Table 8.5) at high concentrations. These 1-mL aliquots can be stored at −20°C but should not be kept longer than 3 months. The aminoglycosides are extremely stable and probably can be held longer than the penicillins, which are more labile (79,87,88). Subsequent dilutions from these buffers are made in appropriate body fluids for assay use.
Antibiotics must be weighed using an analytical balance. The potency of the antibiotic should be calculated based on active micrograms per milligram of powder. In addition, the identical antibiotic must be present in the standard and the sample. For example, gentamicin is a compound composed of three molecular elements. They should be present in the same proportion in the assay standards as they are in the pharmacy.
It is also important that there not be a second antibacterial substance present in the antibiotic standard. Because pharmacy materials may contain preservatives, they should not be used as standards except under emergency circumstances. It is necessary to make standards as close in composition to the patient’s specimen as possible.
For the determination of antibiotic levels in blood, the specimen should be diluted in plasma or serum (89). As discussed subsequently, this principle also applies to the determination of antibiotic levels from other tissue fluids. Considerable work has been performed to establish the types of sera that may be used for dilution. Horse serum, bovine serum, normal human serum, normal human serum inactivated at 56°C for 30 minutes, bovine serum albumin (BSA), and fetal calf serum have been the most extensively studied (61,90–92). It was found that, when antibiotics were suspended in different sera than that used in the test samples, errors of 80% to 367% in measured antibiotic concentrations resulted (6). In assaying antibiotics from cerebrospinal fluid, 150 mmol/L NaCl per 4.5 mmol/L CaCl2 should be added to a phosphate buffer or inaccuracies occur (70). Apparently, it is not necessary to physically buffer the diluent serum (90). It also appears that cations present in sera at physiologic concentrations do not appreciably affect clinical assays (90).
Two factors that are present in sera may, however, exert some effect on microbiologic assays. The gentamicin recovery rate in normal serum has been reported to be between 80% and 90%. However, in uremic serum (blood urea nitrogen levels >50 mg/100 mL), the recovery rate of gentamicin has been shown to be only 50% to 69%. A more drastic decrease in the tobramycin level in the setting of uremia has been demonstrated. There has been some controversy about whether or not high levels of bilirubin in sera can interfere with the results of microbiologic assays of antibiotic concentrations. Blood bilirubin levels of greater than 23 mg/100 mL were shown to cause erroneous determinations of antibiotic concentrations (93). Bilirubin levels of 8 mg/100 mL did not affect the assay under study. Most workers have found that only assays in which bilirubin levels are above 20 mg/100 mL are adversely affected (6,78). These levels are exceedingly rare in clinical samples. Suspending the antibiotic in a matrix equivalent medium that is similar to that from which it came corrects for several factors, the most important of which is protein binding.
Physical Factors
Disks must be known to be effective for assaying antibiotics. Schleicher & Schuell BioScience, Inc (Keene, NH) 740-E disks have been used extensively. Although individual investigators have used disks of different diameters, the smallest disk that produces good zone sizes in the range of the anticipated antibiotic levels should be used. If one is using the recommended 740-E disk, 20 µL of sample should be used in the assay. The maximum volume that this disk accurately holds is 25 µL. The filter paper must lie flat on the surface of the agar or irregular zone sizes will be produced. Supersaturation can lead to surface distortions.
The well technique is approximately five to six times more sensitive than techniques that use paper disks (93). Wells can be conveniently punched in agar with a metal cylinder that is attached to a suction device (94). It is necessary, however, to allow the agar to harden for at least 15 minutes so that cracking does not occur around the wells. The wells can be filled using capillary pipettes because slight overfilling does not produce significant errors in the results obtained (94). Wells should be filled while a low-watt light bulb is maintained at an angle of approximately 30 degrees so that the wells in an assay plate contain uniform amounts of samples or standards. Because the agar depth affects the sensitivity of the test, it is best to add as little agar as possible to the container when a plate is poured.
Choice of Organism
One can use a microbiologic assay to measure the concentration of almost any drug for which a sufficiently susceptible test organism can be isolated (84). It is quite simple to choose an organism for the assay of a sample in which only one antibiotic is present. However, one must exercise considerable caution in organism selection for an assay in which the specimen contains antibiotics in addition to the test drug.
The usual method is to use, as the test organism, a microorganism that is very sensitive to one of the antibiotics and insensitive to the other (81). This dictum appears simple but is often not reliable in practice (61). One must take into account synergy or antagonism, even though the test organism may appear to be resistant to one of the antibiotics. Thresholds at which one antibiotic interferes with another are published (95). The optimum way to assay antibiotics in a mixture is to chemically separate them by electrophoresis or chromatography prior to their measurement (96). This is probably not practical in clinical laboratories. Many test strains have been isolated that adequately take into account the physiologic levels of antibiotic combinations found in humans.
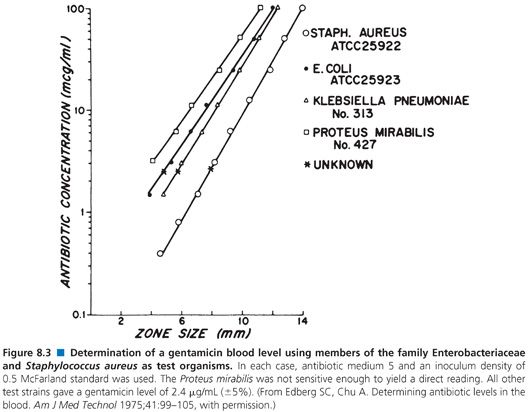
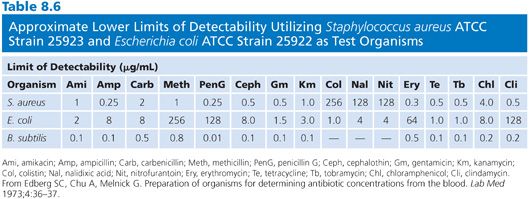
Organisms used for the clinical measurement of antibiotic blood levels most commonly include Bacillus (84,97–100), Staphylococcus aureus (82,93,101,102), Sarcina lutea (103,104), Streptococcus (102,105), Clostridium (99,106,107), Klebsiella, Providencia (108), and others (Fig. 8.3). Fungi and bacteria have been used for the assay of chemotherapeutic drugs (109,110). Assays that use bioluminescent bacteria (111,112) have also been employed. The actual choice of organism depends on the antibiotic to be measured, its susceptibility pattern, its ability to produce clear and crisp zones on the given agar medium, and its ability to provide results within 4 hours. The organism that is selected impacts upon the minimum level of antibiotic that one can detect. Table 8.6 demonstrates the lower levels of detectability for some common antibiotics with assays that use Kirby-Bauer control strains Staphylococcus aureus, American Type Culture Collection (ATCC) strain 25923, and Escherichia coli ATCC strain 25922 as test organisms.
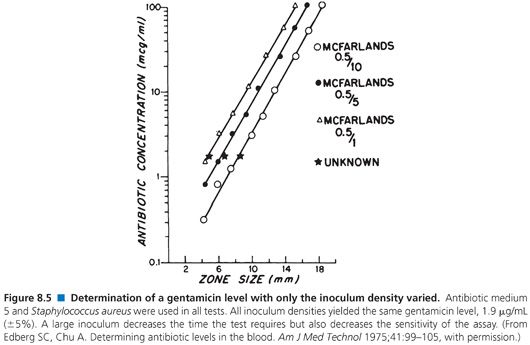
For the rapid determination of antibiotic levels, vegetative organisms must not be more than 24 hours old. The organism can be inoculated the night before the test is run or, more simply, swabbed off a plate, including a Kirby-Bauer sensitivity plate, that is no more than 1 day old (3). An organism with a particularly unusual sensitivity pattern may be seen infrequently. A 4- to 6-hour growth of bacteria can be diluted 1:1 in either fetal calf serum or 7% BSA and stored at −20°C to −70°C for up to 3 weeks. These frozen cultures can be thawed and used directly for rapid antibiotic assays (113,114). Although varying the size of the inoculum changes the sensitivity of the test, this also affects the test time, with a large inoculum reducing both test time and sensitivity.
It is not possible in this chapter to detail all conditions for the assay of the large number of antibiotics that are used in humans. Table 8.7 provides references for assays that are based on the previously mentioned microbiologic principles for some commonly used antibiotics. Table 8.7 also presents the conditions that are required for the determination of concentrations by diffusion assay for some of the major classes of antimicrobials. The choices of test organism, test agar, and buffer diluent shown are optimal for the class of antibiotic. One needs only substitute the required test organism for Klebsiella, use the proper agar and diluent described in Table 8.7, and choose the proper dilution series for the antibiotic standard, as described in Table 8.8.
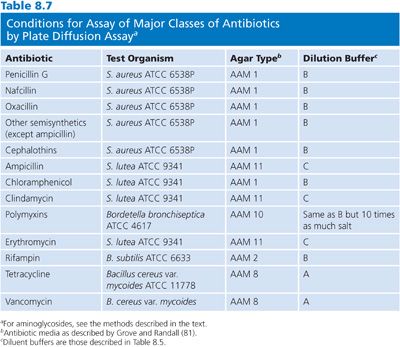
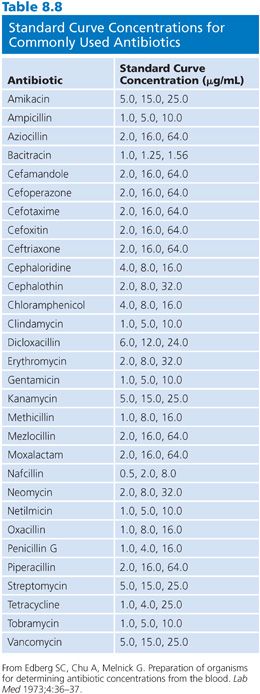
As with other microbiologic assays, when distinct zone sizes appear around the wells or disks, they may be measured. The concentration of antibiotic is calculated in the same way as described for the B. subtilis assay of aminoglycoside concentrations. If one substitutes another test organism, buffer diluent, or test agar for those described here, this would not necessarily invalidate the assay but would likely decrease its sensitivity. As with all assays, one must obtain a straight line for the standards when plotting zone diameters versus logarithms of the antibiotic concentrations. The B. subtilis assay described subsequently can determine the clinically applicable levels of almost all medically used antibiotics (115).
The basic approach used in bioassays is exemplified by the method of Lund et al. (116). This method is typical of those that use organisms that are resistant to all but specific antibiotics for the rapid assay of antibiotic concentrations in clinical material (4,114,117). Although any organism that possesses the appropriate sensitivity and meets the criteria described here can be used, the method of Lund et al. (116) provides a good model. The multiresistant Klebsiella strain they described is available from the ATCC. The method has been successfully field-tested. Prior to the substitution of another organism in this method, users should ensure that the new organism meets the criteria previously described. This strain of Klebsiella is resistant to most commonly used antibiotics, except gentamicin, tobramycin, and amikacin.
Presence of Aminoglycosides
Because the aminoglycoside antibiotics have broad activity, the assay of other classes of antibiotics that are present in combination with them has proved difficult. There are few bacteria that are resistant to aminoglycosides but sensitive to other classes of antibiotics. A group B Streptococcus has been used that allows the determination of clindamycin (CLD) concentrations in the presence of aminoglycosides (118). Care should be exerted when using any streptococcus if a patient is receiving penicillin-type antibiotics because of possible synergy between penicillin and aminoglycosides with this genus. Because aminoglycoside antibiotics are not active under anaerobic conditions, it is possible to determine the level of CLD in the presence of gentamicin using Clostridium perfringens (99). The addition of calcium and other divalent cations to the medium has proved effective (119). However, high levels of calcium may affect bacterial growth and render the medium somewhat turbid. Based on the same principle, cellulose phosphate powder can be incubated with the serum specimen to remove gentamicin (120).
Aminoglycoside antibiotics are inactivated by the polyanionic detergent sodium polyanetholesulfonate in a stoichiometric precipitation reaction (121). The addition of this material to media allows for the rapid determination of the levels of all other classes of antibiotics in the presence of all aminoglycosides (122). Using either nutrient agar or plate count agar, sterile 5% sodium polyanetholesulfonate solution (Grobax; Hoffmann-LaRoche, Nutley, NJ) is added to establish a final concentration of 0.8%. Using the Kirby-Bauer S. aureus ATCC strain 25923, or E. coli ATCC strain 25922, the standard well-type bioassay is run. For penicillin, cephalosporins, erythromycin, tetracycline, CAM, and vancomycin, S. aureus is the better choice. For antibiotics that are more active against gram-negative bacteria, such as ampicillin or the polymyxins, E. coli is preferred. The procedure described for the Klebsiella assay can be followed with either of these strains.
One-Dimensional Assays
As previously described, one-dimensional assays can be useful for the determination of antibiotic concentrations under anaerobic conditions and for assays for which only small amounts of specimen are available.
Medium
Nutrient agar is diluted with an equal amount of 1% peptone in water and brought to pH 7.8 (the pH varies depending on the type of antibiotic to be assayed). It is dispensed for storage in 19-mL amounts.
Procedure
The bacterial test strain (S. aureus 658P) is grown overnight in trypticase soy broth. It is diluted 1:100 and mixed well by shaking to break up clumps. Before use, the agar is melted and cooled to 48°C in a temperature-controlled water bath. Then 1 mL of the bacterial suspension is added to 19 mL of the test agar. The bacteria and the test agar are mixed well. The test agar is pipetted into conical test tubes that have an internal diameter of approximately 3 mm (K tubes used for determination of complement fixation in the Kolmer test in syphilis serology are satisfactory). The agar is added to a depth of 3 mm from the bottom of the tube. The agar is allowed to harden (at least 5 minutes). Standards for many commonly used antibiotics are described in Table 8.5. Each standard is pipetted into a different tube. Each standard should be repeated three times. The patient’s serum is treated identically. The antibiotic standards and patient’s serum should be added until they are 1.5 mm above the agar layers. Small deviations from this level do not affect the accuracy of the method. The tubes are incubated at 37°C overnight.
Interpretation
Typical assay results are presented in Figure 8.1. To determine inhibition of growth, each tube is laid on its side. Using an eyepiece with the ability to measure distances, the distance in millimeters from the point where the patient’s specimen and the test agar meet (the meniscus of the test agar) to the point where growth of the test strain begins (often it is the place where large colonies are seen) is recorded. This measurement is repeated for each standard and the patient’s specimen. The results are calculated as for any dose-response curve. On the ordinate (y axis), the logarithm of the antibiotic concentration is plotted. The depth of inhibition is plotted on the abscissa (x axis). The concentration of the patient’s specimen is found by drawing a vertical line from the depth of inhibition on the x axis to the standard line and drawing a horizontal line from the point of intersection to the y axis, where the concentration of the antibiotic is read directly.
Amoxicillin and Clavulanic Acid
β-Lactamase inhibitors protect β-lactam antibiotics from destruction by these enzymes. Clavulanic acid, sulbactam, and tazobactam are those currently used. Because β-lactamase inhibitors are combined with β-lactam antibiotics in the same pharmaceutical preparations, a procedure for the assay of amoxicillin and clavulanic acids is presented (123).
Augmentin consists of amoxicillin trihydrate and the potassium salt of clavulanic acid as the anhydrous free acids in the ratio of two parts amoxicillin to one part clavulanic acid. Amoxicillin may be assayed in the presence of clavulanic acid. A conventional microbiologic assay technique with S. lutea as the assay organism can be used to measure amoxicillin concentrations in body fluids after the administration of Augmentin. This is not the case with clavulanic acid because its very low level of antibacterial activity precludes the use of a microbiologic assay technique that depends on measurement of antibacterial activity. Instead, clavulanic acid concentrations in body fluids can be assayed by a microbiologic agar diffusion method involving measurement of the inhibition of β-lactamase activity of a strain of Enterobacter aerogenes. In the method subsequently described, clavulanic acid does not interfere with the microbiologic assay of amoxicillin in clinical specimens, and amoxicillin does not influence the measurement of clavulanic acid.
This method uses a large-plate microbiologic assay technique in which samples are added to wells punched in agar. Modifications may be made as required; for example, Petri dishes may be used instead of large plates, and the specimens may be applied to the plates by assay cylinders, paper disks’ fish spines, beads, and others. Moreover, the bacteriologic media used are not critical and individual laboratories may find their own modifications to be more suitable for their purposes than those described.
In principle, specimens should be assayed as soon as possible after collection and should not be stored for more than a few days before assays are performed. As a rule, β-lactam compounds are relatively unstable in aqueous solutions and in body fluids, and clavulanic acid is no exception. Consequently, care must be taken in the handling and storage of clinical specimens that contain amoxicillin and clavulanic acid. Serum specimens may be stored for up to 2 days in the refrigerator (4°C) or for up to 3 days in a freezer (−20°C). Clavulanic acid is relatively unstable in undiluted urine, particularly at alkaline pH, and is less stable (in urine) at −20°C than at 4°C. Accordingly, urine specimens for assay should be diluted 10-fold in citrate buffer, pH 6.5, as soon as possible after collection and stored at 4°C. Under these conditions, specimens of urine containing amoxicillin and clavulanic acid may be kept for up to 5 days at 4°C before assay.
The stability of clavulanic acid is influenced by the concentration, pH, and composition of the buffer solution that is used as a diluent. Solutions of clavulanic acid are most stable at pH 6.0 to 7.0, and preparations in citrate buffers or distilled water are more stable than those in phosphate buffers. Also, aqueous solutions that contain amoxicillin and clavulanic acid are more stable at 4°C than at −20°C. Consequently, aqueous solutions of Augmentin, clavulanic acid, or amoxicillin for microbiologic tests should be prepared in 0.1 mol/L of a citrate buffer, pH 6.5. Solutions that are prepared in this medium and contain up to 0.1 mol/L of amoxicillin or clavulanic acid may be stored at 4°C for up to 4 weeks.
Amoxicillin
Apparatus. The following equipment are required:
1. Large glass assay plates
2. Pasteur pipettes (nominally 30 drops/mL) or standard dropping pipettes (0.02 mL per drop)
3. Punches for cutting holes (7- to 8-mm diameter)
4. Lancets or broad needles
5. Test tubes, flasks, and pipettes as required
6. Leveling tripods or a level surface
7. Water bath at 50°C
8. Incubator at 30°C or 37°C
9. Needle-point calipers
Antibiotic medium 2 (81) is obtainable commercially in dehydrated form from Chesapeake Biological Laboratories (Baltimore, MD), Oxoid, Inc (Ogdensburg, NY), and Difco Laboratories (Sparks, MD). It is prepared by dissolving beef extract (1.5 g), yeast extract (3.0 g), peptone (6.0 g), and agar (15.0 g) in 1,000 mL of distilled water and adjusting the pH to 6.5 or 6.6. Volumes of 300 mL are distributed in screw-capped bottles and sterilized at 15 psi for 15 minutes.
The required amount of medium is melted and maintained at 50°C in a water bath until the plates are ready to be poured. The assay plates are placed on a level surface or on leveling tripods and adjusted until they are level. The plates are sterilized by swabbing them with alcohol, followed by flaming. The swabbing and flaming procedure is performed twice. The inoculum is added to each bottle of agar and mixed thoroughly. The surface of the plate is flamed and the inoculated medium is poured evenly over the plate. The surface of the agar is again flamed to eliminate air bubbles, and the agar is allowed to solidify with the lid slightly open. The plates may be kept in a refrigerator at 4°C for up to 24 hours.
Buffer. Sorensen’s buffer (0.1 mol/L citrate buffer, pH 6.5) is used. Solution A is 0.1 mol/L disodium citrate (21.0 g of citrate acid in water, dissolved in 200 mL of 1 N NaOH [4.0 g/1,000 mL] and diluted to 1,000 mL). Solution B is 0.1 N NaOH (4.0 g/1,000 mL). Fifty-four milliliters of solution A is added to 46 mL of solution B. The pH of final solution is checked and, if necessary, adjusted by adding the required amounts of solution A or B.
Standard Solutions. Normal pooled human serum is sterilized by filtration and tested for the absence of antibacterial activity. The quantity of laboratory reference standard amoxicillin trihydrate that is equivalent to the required amount of amoxicillin pure, free acid is accurately weighed and dissolved in 0.1 mol/L citrate buffer, pH 6.5. Dilutions are made in pooled human serum for the assay of serum specimens or in 0.1 mol/L of a citrate buffer, pH 6.5, for the assay of other specimens or urine. Serial dilutions are prepared in the requisite diluent to give the necessary range of standard solutions. Each specimen is diluted to give a concentration that is estimated to fall within the range of the standard line.
Serum specimens are diluted in pooled human serum. Specimens of urine are diluted in 0.1 mol/L citrate buffer, pH 6.5. The required dilution depends on the dose of the drug that was administered and on the time at which the specimen was taken. The mean serum concentrations of amoxicillin and clavulanic acid in fasting volunteer subjects 1 hour after the administration of single oral 375 mg doses of Augmentin are 5.6 µg/mL for amoxicillin and 3.7 µg/mL for clavulanic acid. The mean urine concentration of clavulanic acid is 545 µg/mL.
Bacteria. A suspension of S. lutea (National Collection of Type Cultures strain 8340, ATCC strain 9341) is prepared by using a loop to inoculate 100 mL of nutrient broth in a 500 mL Erlenmeyer flask with organisms from a stock culture that has been grown on a nutrient agar slant and stored in the refrigerator at 4°C. The flask is incubated for 48 hours at 37°C and stored at 4°C. Stock cultures on agar slants may be kept for up to 4 months at 4°C. Broth suspensions may be held for 4 or 5 weeks at 4°C. An inoculum of 0.8 mL of the broth suspension into 100 mL of agar usually provides satisfactory growth on the large assay plates.
Procedure. Seeded plates are taken from the refrigerator and the agar surface is blotted dry with filter paper. Holes 8 mm in diameter are cut with a punch. The agar plugs are removed with a lancet or broad needle. The holes are filled with the specimens and standard solutions using a Pasteur or dropping pipette. The pipette is rinsed three times in buffer solution between each sample loading. The plates are incubated overnight at 30°C or 37°C.
Interpretation. The diameters of the inhibition zones are measured and the standard and sample responses are averaged. The mean inhibition zone diameters of the standard solutions are plotted against the logarithm of the antibiotic concentrations using semilogarithmic paper. The best fitting straight line connecting the points is constructed. The concentration of each dilution of specimen is determined by extrapolation from the straight line.
Clavulanic Acid
Clavulanic acid is a weak antibacterial agent. However, it is a potent inhibitor of certain β-lactamases. This latter property is the basis of a microbiologic assay for clavulanic acid in clinical specimens. In brief, a subinhibitory concentration (60 µg/mL) of benzylpenicillin is added to a nutrient agar that is inoculated with the β-lactamase–producing organism E. aerogenes BRL strain 1. Plates are poured and wells are cut in the agar in the usual fashion. At the concentrations tested, clavulanic acid has no inhibitory effect on the growth of the assay organism, but it does inhibit the β-lactamase activity of the enterobacterium, thereby preventing destruction of the benzylpenicillin incorporated in the agar. As a consequence, inhibition zones are produced by the penicillin, the diameters of which are proportional to the concentration of clavulanic acid in the test sample.
The assay for the determination of clavulanic acid is identical to that for amoxicillin, with the following exceptions. The assay medium is adjusted to pH 7.4. The amount of laboratory standard material of potassium clavulanate that is equivalent to the required amount of the pure, free clavulanic acid is accurately weighed and dissolved in 0.1 mol/L of a citrate buffer, pH 6.5. Nutrient broth inoculated with a wire loop from a nutrient agar slant of E. aerogenes BRL strain 1 (or Klebsiella pneumoniae ATCC strain 29665) and incubated overnight at 37°C is used to inoculate the assay agar. A fresh culture should be used for each assay. An inoculum of 3.0 mL of overnight broth culture in 100 mL of agar produces satisfactory growth on large assay plates.
The inoculum is added to the agar and benzylpenicillin is added to the inoculated agar to give a final concentration of 6 µg of benzylpenicillin per milliliter of agar. The agar is poured into the plates, as described for the assay of amoxicillin. When the agar has set, the plates are stored in the refrigerator at 4°C and are used as soon as possible on the same day. This is essential because the assay organism is able to inactivate the penicillin incorporated in agar plates if they are allowed to stand at room temperature or overnight at 4°C. The results are interpreted as for the amoxicillin assay as previously described.
Three-Dimensional Assays
Antibiotic medium 11 (Difco Laboratories, Detroit, MI) is adjusted to pH 7.9. This agar may be stored in 25-mL aliquots in the refrigerator for up to 1 month. Prior to performing the assay, an appropriate number of tubes are melted and brought to 50°C; 0.4 mL of an overnight growth of Klebsiella in trypticase soy broth is added. The suspension is thoroughly mixed and 9 mL is poured into two 100 × 15-mm plastic Petri plates. Alternatively, 0.4 mL of a heavily inoculated 5-hour broth culture can be added to the agar.
Procedure
For each assay, 0.02 mL of the patient’s serum is pipetted with sterile disposable capillary pipettes onto Schleicher & Schuell 740-E disks (Schleicher & Schuell BioScience, Inc, Keene, NH). Alternatively, wells can be cut in the agar. The method described by Sabath et al. (6,93) is followed using the same number of standards and samples of the patient’s serum.
Calculation of Results
Generally for a given antibiotic, results are obtained from a plot of the logarithm of the antibiotic concentration versus the zone diameter. This is quite adequate for calculations involving limited ranges of antibiotic concentrations. However, if the concentration range is greater than fourfold, it is better to plot the logarithm of the antibiotic concentration versus the square of the zone diameter. Lines that visually appear straight on plots of the logarithm of the antibiotic concentration versus zone diameter have frequently been shown by computer analysis to have low coefficients of variation (97). This caveat is particularly important if the slope of a line is steep. When using logarithmic paper to calculate antibiotic concentrations in this way, very small differences in values can produce large changes in the apparent concentrations, especially in the range of 10 to 100 µg/mL. It is best to use an assay system that provides the flattest possible line. Pocket calculators or handheld computers can provide “best fit” straight lines, coefficients of variation, SDs, and direct interpolations. Keeping permanent records of these calculations provides a strong internal quality control. One may, for example, detect the decay of antibiotic standards by changes in line slopes or differences in coefficients of variation.
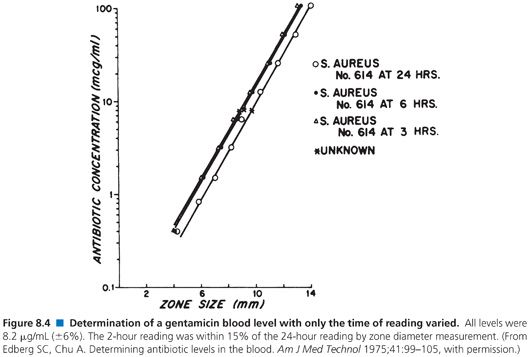
When used to perform clinical assays, the microbiologic method produces results in 4 hours or less. Figures 8.4 and 8.5 illustrate that longer incubations affect the zone size only slightly. The size of the zone is, in effect, established approximately 2 or 3 hours after incubation. Small, uniform increases in zone size do not affect the slope of the line or the final calculation of antibiotic concentration.
Electrophoretic Separation of Antibiotics
Methods have been developed to separate antibiotics in a mixture by gel electrophoresis and to assay these separated compounds by covering the gel with an indicator bacterium in agar. After incubation, zones of inhibition are seen in the covering agar slab. These methods were especially useful before HPLC became available and may still have some applicability if one needs to assay for disparate classes of antibiotics in a mixture (124,125).
Broth Methods
Turbidimetric Assays
For completeness, the basic parameters of the assay of antibiotics by turbidimetric means are mentioned. These techniques have largely been relegated to the assay of antibiotic-containing materials in situations in which one wants to screen large numbers of samples in a short period of time. Turbidity is used here to mean any technique in which the growth of bacteria in a liquid medium is used to quantify the amount of antibiotic in a solution. Included under this heading is a consideration of techniques in which the change in the number of bacteria is directly measured photometrically (as numbers or mass of organisms) and, as an extension, those in which a product such as a change in pH or the release of CO2 is measured.
An examination of the theory of optical methods was explored by Kavanaugh (79). Only the pertinent factors involved in the assay of antibiotics by turbidimetric means are discussed here. The assay of antibiotics by photometry depends on the direct relationship between growth of bacteria in culture and the amount of antibiotic present. Generally, this relationship is displayed in a plot of growth per unit time versus percent transmission or the optical density of the bacterial suspension. It should be noted that absorbance measures mass or volume rather than the concentration of bacteria (79). This relationship can be stated as a form of Beer’s Law or Beer-Lambert Law in which Io is the amount of light leaving a suspension, I is the amount of light entering a suspension, OD is the optical density, N is the mass of bacteria, E is the extinction coefficient of the particles, a is the optically effective area of the particle, and b is the thickness of the suspension. Early work on the assay of antibiotics utilized modifications of this equation from commercial fermentation processes for the assay of antibiotics. Several investigators have attempted to modify it for use in clinical assays (105).
log(Io/I) = OD = (N)[Eab/2.3]
Irrespective of the instrument or technique used, procedures must be employed to straighten the generally curved calibration lines for turbidimetric microbiologic assays. Although it is beyond the scope of this chapter to treat the subject fully, the following relationships should be useful for the applied assay. A theoretical dose-response line for an antibiotic that decreases the growth rate of the test organism is
Nt = No exp(ko + f(v)kM – kaC)t,
where Nt is the concentration of the test inoculum after an incubation period of t, No is the concentration of the test inoculum at the beginning of the assay, ko is the generation rate constant in the absence of antibiotic, C is the concentration of antibiotic with an inhibitory coefficient of ka, kM is the effect of the medium, and the function f(v) is a function of the volume of the sample added to the assay. This is a theoretical expression because the major characteristics of the equation, although interrelated, are not absolutely known.
In an individual assay, No, ko, f(v), kM, ka, and t (time) are constant. Therefore, the variables of the equation may be related as follows:
log N = G + BC,
where G and B are constants that are intrinsic to the assay. Because most spectrophotometers use absorbance (A), this equation may be translated into
log A = E + FC.
To straighten the line, this equation may be modified as
log (A + M) = O + PC.
The algebraic sign M is used to straighten the line over the particular concentration range in question. The constant M compensates for two sources of curvature: the nonlinear relationship between log N and C, and the inherent nonlinear responsiveness of photometers.
Because of this complexity, pure photometry has proven successful only in situations in which all conditions except the quantity of a single antibiotic in solution could be rigidly controlled. The Abbott Laboratories MS-2 instrument (Abbott Park, IL), a device that continually monitors bacterial growth in optical density units, has been used to determine antibiotic blood levels under clinical conditions using a growth curve analysis (126). The principle of this technique is that, for any dose-response curve, the response is the turbidity of bacteria in solution. The MS-2 instrument determines turbidity using red light–emitting diodes. The test culture is inoculated into the cuvette. After the culture reaches logarithmic growth phase, the culture is pulled down into compartments, each of which contains a disk. The disks can contain either a patient’s serum or a standard. Generally, the patient’s serum is tested in two compartments and each standard is repeated twice. After approximately 3 hours of incubation, the optical density (after minor corrections that are required because of the physical nature of the instrument) is plotted on the y axis and the logarithm of the antibiotic concentration is plotted on the x axis. The instrument has a built-in computer that enables it to continuously monitor the growth of the test cultures. All calculations are automated. In the early 1980s, we used this instrument successfully with S. aureus as a test organism to determine the concentration of aminoglycosides in blood (126).
Broth Dilution Bioassay for Polymyxins
Broth dilution bioassay is rarely performed. However, this technique may prove useful for the measurement of polymyxin-class antibiotics. It was found that in normal agar, but not agarose, assays of colistin concentration yielded false-low blood and urine values (127). This was probably occurring because a highly diffusible precursor was being converted into a poorly diffusible active compound. The turbidimetric technique more accurately reflected what was actually occurring in the body than did agar techniques. Levels of the polymyxin group of antibiotics can be determined by a modification of the standard turbidimetric technique in which the number of viable organisms is counted.
Procedure. A strain of Pseudomonas aeruginosa that is sensitive to the polymyxins is employed as the test organism. The inoculum that is used for each antibiotic concentration determination is 0.05 mL of a 1- to 8-hour culture (grown in trypticase soy broth) that has been diluted at a 1:10,000 ratio in normal human serum. The patient’s specimen is diluted in the same fluid that is to be assayed (serum specimens are diluted in serum, cerebrospinal fluid specimens are diluted in cerebrospinal fluid, and so on) to yield final percentage concentrations of 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 10%, and 0%. The final volume of each of these dilutions is 0.5 mL after the addition of 0.05 mL of the inoculum. Standards should be incubated with normal human serum for 18 hours at 37°C. This preincubation is necessary for activation of the polymyxin-type drugs.
Immediately after the addition of the inoculum, all tubes are incubated at 37°C for 30 minutes. After incubation, the tubes are plunged into an ice bath (0°C). The number of bacteria in each tube is determined by performing serial 10-fold dilutions with distilled water and plating 0.1 mL of each dilution on the surface of trypticase soy agar plates. The total number of viable bacteria per milliliter of inoculum is calculated from these colony counts after 18 hours of incubation at 37°C.
The end point for each titration of the polymyxins in each patient’s serum (or other fluid) is the smallest amount of patient’s serum that results in a reduction of inoculum to 10% of the original viable count. To increase the precision of the end points, the concentration of the subject’s serum or the concentration of known antibiotic that causes a reduction of 90% of the viable organisms is read from a curve that is drawn on semilogarithmic paper by plotting the number of organisms on the logarithmic axis versus the concentration on the arithmetic axis. Performing the assay with normal human serum alone accounts for any reduction in bacteria that is caused by nonspecific factors.
Interpretation. The antibacterial activity of the polymyxin standards is compared with the antibacterial activity of the patient’s specimen. The calculations are as set forth in the “Turbidimetric Assays” section. The titer of the patient’s serum is multiplied by the minimum inhibitory concentration of the organism for the individual polymyxin to yield the amount of polymyxin-type antibiotic in the patient’s serum.
pH Change Assays
Antibiotics act on bacteria by halting their growth and/or metabolism. Antibiotic-induced changes bacterial metabolic activity can be used to determine antibiotic concentrations in body fluids and tissues. The methods used are based on the principle that the larger the quantity of antibiotic that is in contact with a bacterial population, the greater will be the modification of the population’s metabolic pathways. As with all dose-response assays, one must obtain a standard dose-response curve and calculate from only those points that fit the equation for a straight line.
The most popular method of this type employs a change in the pH of the growth medium as a measure of the amount of antibiotic that is present. If no antibiotic is present, the test strain grows and produces a change in the pH of the growth medium by metabolizing a constituent (e.g., a sugar, which would lower the pH, or urea, which would increase the pH). If an antibiotic is added to the system and the test organism is affected by it, the change in pH is lessened. The urease-based bioassay is representative of this group of assays and can be used by laboratories without special equipment.
Adenosine Triphosphate Measurement Assays
A potentially automatable assay that is a combination of a bioluminescence procedure and a chemical assay uses rates of endogenous adenosine triphosphate (ATP) production by Klebsiella edwardsii to determine antibiotic concentrations. The amount of ATP that is present in a broth reflects bacterial growth, which is inversely related to the amount of antibiotic present. Standards are inoculated with the test strain in parallel with the patient’s serum. After 2 hours of incubation at 37°C, the tubes are extracted with an ethylenediaminetetraacetate/H2SO4 solution. The amount of bacterial ATP that is released is determined by spectrophotometry (128). A plot of the antibiotic concentration versus the relative amount of ATP released is used to calculate the antibiotic blood level. One should heat the serum prior to performing the assay in order to destroy human adenosine triphosphatase. Instruments that enumerate bacteria in urine and other body sites based on this principle are available from a number of vendors, including Celsis International (Newmarket, Suffolk, United Kingdom), Coral Biotechnology (San Diego, CA), and New Horizons Diagnostics Corporation (Columbia, MD).
Bioluminescence
Methods that involve the use of luminescent bacteria have been proposed to determine the activity of antibiotics in serum (111,112). One type of assay called the induced test is based on the ability of some antibiotics to inhibit luciferase synthesis by luminescent bacteria (129). Ulitzur’s (129) method, described in the following section, is sensitive, rapid, and potentially automatable.
Procedure. The serum is heated at 56°C for 30 minutes to eliminate bacterial activity. To 0.8 mL of serum, 0.2 mL of 10% NaCl containing 0.1 mol/L 3-(N-morpholino) propanesulfonic acid buffer and 1.5% glycerol is added. The final pH value of the mixture should be 7.9 for all antibiotics except CAM and tetracycline, which are tested at pH 6. A Photobacterium leiognathi 8SD18 cell suspension (200 µL, 3 × 108 cells/mL) is added to 0.8 mL of the serum, as well as to 1 mL of antibiotic-free pooled serum. After 10 minutes of preincubation at 30°C, proflavin is added to give a final concentration of 1.5 µg/mL for pH 7.9 or 25 µg/mL for pH 6.0. The vials are then incubated with gentle shaking at 30°C. Proflavin is a DNA-intercalating agent that induces the luminescence system of dark mutant luminescent bacteria. The results are recorded after 40 to 60 minutes of incubation. Luminescence is measured with a photometer/photomultiplier and is universely proportional to the concentration of the antibiotic in the specimen.
The bioluminescence test specifically determines the activity of the tested antibiotic as a de novo protein synthesis inhibitor. Antibiotics that act on DNA or cell wall synthesis are not detected by this test. The bioluminescence test is more sensitive than most available bioassays. This high sensitivity may be attributable to the greater susceptibility of newly synthesized proteins to the inhibitory actions of many antibiotics.
Serum Inhibitory Concentration and Serum Bactericidal Concentration
Broth dilution tests that employ patient serum as the antimicrobial milieu date to the work of Schlichter and McLean (130), who reported on the serum inhibitory concentrations (SIC) in 10 patients with streptococcal endocarditis. They obtained serum from these 10 patients, geometrically diluted each sample in broth, and inoculated each dilution with the patient’s own organism (131). They obtained a titer of each patient’s serum that reflected the serum’s ability to inhibit macroscopic growth of the microbe. Fisher (132) subsequently extended this procedure by subculturing each tube that failed to exhibit macroscopic evidence of growth after overnight incubation. The highest dilution of a patient’s serum, or titer, that was able to kill the microbial inoculum was called the serum bactericidal concentration (SBC). SIC/SBC tests have since been applied to specimens from patients receiving therapy for intravascular and other closed-space infections (133).
Procedure. The patient’s infecting microbe is adjusted to yield between 105 and 106 colony-forming units (CFU)/mL in each tube (134). It is important that the actual CFU per milliliter value be determined. To do this, 0.1 mL each of a 1:100 and a 1:1,000 dilution of the inoculum is spread over the surface of the appropriate agar medium. After overnight incubation, the inoculum size is calculated from the number of colonies that are present on the plate that has between 20 and 200 colonies on it. For example, if there are 40 colonies on the plate that was inoculated with a 1:1,000 dilution, the original inoculum contained 4 × 105 CFU/mL.
The serum is diluted geometrically and the test is performed in a manner that is analogous to the minimum inhibitory concentration/minimum bactericidal concentration procedures. For most organisms, the broth can be Mueller-Hinton broth, with brain-heart or Levinthal medium used for fastidious microbes. To each of 12 tubes, 1 mL of broth is added. To the first tube is added 1 mL of serum, followed by thorough mixing. One milliliter from tube 1 is added to tube 2 and is thoroughly mixed. One milliliter from tube 2 is added to tube 3, and so forth. This process is continued through tube 12. However, 2 mL of broth is added to tube 12, rather than 1 mL as in the other tubes. One milliliter is removed from tube 12 and is transferred to a sterile test tube as a sterility control. A geometric dilution of the patient’s serum has, therefore, been made from 21 to 212. One should also have a 14th tube that contains only the patient’s undiluted serum. To each tube is added 1 mL of the patient’s own microbe, diluted to yield a final concentration of 105 or 106 CFU/mL. All transfers should be made with sterile pipettes. All tubes are incubated in ambient air at 35°C for 18 to 24 hours. The lowest concentration (dilution) of the patient’s serum that completely inhibits visible growth is the SIC. Concentration or dilution is converted to titer by the formula: titer = 1 per dilution. As the titer increases, the amount of serum in the tube decreases.
The SBC is determined by spreading 0.1 mL from each tube that does not show turbidity and the first tube that does show turbidity over the entire surface of a 100-mm diameter agar plate that contains the appropriate growth medium. A different pipette should be used for each transfer from a tube. The plates are incubated for 24 hours and preliminary colony counts are performed and recorded. The plates should be incubated for 48 hours when examining staphylococci and for up to 72 hours when examining other microbes. This extended incubation allows the organism to grow on the surface of agar and obviates any effects that transferred antibiotic may exert. The SBC is the lowest concentration (i.e., highest titer) of serum that produces 99.9% killing. For example, if the inoculum in each tube was 2.0 × 105 CFU/mL, killing would be defined as an agar plate that demonstrates no more than 20 colonies from a 0.1 mL subculture.
Interpretation. Much controversy surrounds the use of SIC/SBC tests (135,136). Importantly, few groups have performed the procedure in the same manner. Variations in protocols have involved inoculum size, type of broth, bactericidal end point, time of incubation, timing of the blood sample, volume of the broth sample, and the definition of the bactericidal end point. For these reasons, this test is no longer performed at Yale.
DIRECT CHEMICAL ASSAYS
Although considerable effort has gone into the determination of antibiotic concentrations in body fluids by direct chemical analysis, there are few instances in the clinical laboratory when one can use these methods. Because these assays are based on reactions that involve specific chemical groups on antibiotic molecules, the concentrations of antibiotics determined by many chemical assays include active drugs as well as metabolic breakdown products that retain the reacting moiety. Consequently, a chemical assay is most satisfactory in a pharmaceutical fermentation process in which one is dealing with a pure substance.
In vivo, from 0% to more than 95% of an administered drug may not exist in its native state. Because a clinical assay is intended to determine the amount of active material circulating, chemical assays may yield false-high values. In addition, for many antibiotics, all their breakdown products may not be known. When chemically assaying the amount of active drug from biologic sources for such antibiotics, it is dangerous to use estimates of average metabolically active fractions. In addition, secondary substances (either other chemotherapeutic agents or normal body constituents) may interfere with these assays. Because it is very difficult to exclude all possible agents in a given assay, falsely elevated values can unpredictably result.
Most chemical assays require extraction of the antibiotic prior to analysis. This extraction leads both to an inordinate number of technical procedures and to a greater possibility of error. Also, extracted material often requires specialized environmental safety conditions. Although these obstacles are not insurmountable in industry, they may be unwieldy for clinical laboratories.
The advantages of chemical assays for clinical laboratories are, at present, more theoretical than practical. Chemical assays can potentially be automated and should provide rapid turnaround times. Moreover, chemical analysis yields an absolute quantity, as opposed to a relative response, as in the microbiologic assay. This makes the standardization and implementation of controls easier. Only those assays that are useful in the routine and/or clinical research laboratory are discussed in the following paragraphs.
Colorimetric Assays
Aminoglycosides/Aminocyclitols
The thiobarbituric assay of streptomycin has been modified to allow it to determine clinically important levels of many deoxy sugars (137). Although the chromatogen that is produced when streptomycin and thiobarbituric acid react is quite stable, biologic homogenates that contain glycoproteins, sugars, and plasma proteins have been found to interfere with the assay. Changing the temperature of incubation from 100°C to 37°C eliminates all but plasma protein interference. Automated dialysis and computerized result analysis can potentially be used to design automated assays based on the method.
A crude solution of the enzyme is used in the reaction. The acetylated product, rather than being adsorbed to phosphocellulose paper, is not used at all. Thiol coenzyme A that is produced in the reaction is allowed to react with 5,5′-dithiobis(2-nitrobenzoic acid). The product of this reaction is thionitrobenzoic acid, a compound that has a maximum absorbance at 412 nm. The amount of thionitrobenzoic acid produced is a measure of the aminoglycoside concentration. For successful application of this assay, protein impurities must be removed. One should use a more pure enzyme suspension than that required for radioenzymatic assay procedures. With this assay system, gentamicin blood levels can be obtained within 15 minutes (138). Efforts to increase the sensitivity of the assay and to establish the optimal reaction conditions are underway. It should be noted that vancomycin, which is often used in conjunction with gentamicin and streptomycin, does not interfere in the chemical assays described in this chapter.
β-Lactam Antibiotics
Concentrations of the penicillins and related antibiotics in human specimens have been spectrophotometrically determined by a variety of methods. Most methods rely on a β-lactamase to hydrolyze the β-lactam ring of the drug and involve the measurement of the end products of the reaction (139,140). Historically, these methods have suffered from an inability to distinguish one form of penicillin from another. A relationship can be established between the consumption of iodine by a penicillin and the quantity of the penicillin in solution.
In one useful method, interfering protein is easily removed. Isopropyl alcohol is added to an aliquot of serum to remove the protein. The protein is precipitated by centrifugation (16,000 rpm in a Sorvall RC-2 centrifuge [Thermo Fischer Scientific, Inc., Waltham, MA, Norwalk, CT]). Two equal volume aliquots of the supernatant are taken. One aliquot is used as the sample, and the other is used as a blank. Iodine solution (0.01 N), buffered at pH 6.5, is added to each aliquot. A measured amount of aqueous penicillinase (Rikker penicillinase, 1,000 units/mL) is added to the sample while an equal amount of distilled water is added to the blank. After 25 minutes, the excess iodine in each tube is quantitated using a thiosulfate reagent and a starch indicator. The penicillin concentration can be calculated from the difference in iodine uptake between the specimen and the blank. The amounts of base and penicillinase that are required, as well as the incubation times, vary with the penicillins that are being measured. As a general formula, the amount of penicillin-type antibiotic (although susceptibility to penicillinase varies) can be calculated by the general formula:
µg/mL penicillin = (V2−V1)(MW/N)/sample weight in mg
where V2 is the volume of 0.01 N iodine consumed after inactivation with alkali, V1 is the iodine consumption before inactivation, MW is the molecular weight of the individual penicillin, and N is the number of iodine equivalents (141).
Another technique is to assay for a specific constituent of the penicillin molecule. This approach proved successful in the determination of 6-amino-penicillinoic acid with a glucosamine reagent (142). An additional method of analysis relies on the differential absorption of light in a given wavelength by different penicillin molecules to determine antibiotic levels when more than one drug is present. Ampicillin has a higher absorbance at 268 nm in a solution of pH 5 than in one at pH 9. This property has been used to measure concentrations of ampicillin in the presence of cloxacillin (143).
To avoid the difficulties inherent in the alkalized starch-iodine method, a procedure was developed that substituted chloroplatinic acid for base in the assay. Chloroplatinic acid degrades penicillin to penicillinoic acid, which can be measured colorimetrically. The color generated is more stable than that produced with the alkaline method. In the assay, 0.13 mL of 0.2% chloroplatinic acid is added to the penicillin-containing specimen and brought to a final volume of 4 mL with distilled water. The mixture is kept at room temperature for 30 minutes, after which time 1.5 mL of starch-iodine color reagent is added. The starch-iodine color reagent is prepared by adding equal volumes of water-soluble starch solution (0.8%) and 480 mol/L of a 4.8 mmol/L potassium iodine solution. After incubation for 5 minutes at room temperature, the amount of penicillin may be calculated from the absorbance measurement at 260 nm (101).
Active Sulfonamides
The body fluid is extracted into ethyl acetate from a nondeproteinized sample, yielding active unchanged sulfonamide and an acetylated inactive component. The acetylated component does not react in this assay.
Reagents. The following reagents are used:
1. McIlvain buffer, pH 5.5, which is made by mixing 8.6 volumes of a 0.2 mol/L aqueous solution of citric acid with 11.4 volumes of a 0.4 mol/L aqueous solution of disodium phosphate
2. Ethyl acetate
3. 2 N solution of HCl in acetone/water. The required quantity of this reagent must be freshly prepared immediately before each series of analyses by mixing one volume of 8 N HCl with three volumes of acetone. This product should not be kept for more than a few hours and should be discarded as soon as a brown color develops.
4. 0.1% sodium nitrate in a mixture of 3:1 acetone/distilled water
5. 5% solution of sulfaminic acid in 3:1 acetone/distilled water
6. 0.1% solution of α-naphthylethylenediamine dihydrochloride in a 3:1 mixture of acetone/distilled water
7. Methanol
Procedure. For plasma samples, 1 mL of McIlvain buffer is pipetted into a 10- to 15-mL shaking tube that is sealed by either an ether-tight glass or polyethylene stopper. Plasma (0.1 mL) and ethyl acetate (5 mL) are added. The tube is mixed by shaking for 10 minutes for extraction and simultaneous partial deproteinization and centrifuged 5 minutes at 3,000 rpm. The proteins settle between the two liquid phases as a fine precipitate.
Three milliliters of the supernatant is transferred to a test tube and 0.5 mL of HCl is added and mixed. Then 0.5 mL of 0.1% sodium nitrite solution is added, mixed, and allowed to stand for 6 minutes. One-half milliliter of sulfaminic acid solution is added and mixed well by shaking until there is no further liberation of gas bubbles. After 3 minutes, 0.5 mL of α-naphthylethylenediamine dihydrochloride solution is added and mixed. One-half milliliter of absolute methanol is then added. The tube is mixed until a homogeneous liquid phase is achieved. The tube is closed with a polyethylene stopper. In 20 minutes to 1 hour, one can determine the concentration of the product photometrically, as described in the following texts.
Total Sulfonamides
Reagents. The following reagents are used:
1. 20% trichloroacetic acid
2. 3 N aqueous HCl
3. 0.1% aqueous sodium nitrite
4. 0.5% aqueous sulfaminic acid
5. 0.1% aqueous solution of α-naphthylethylenediamine dihydrochloride (Note that all reagents can be stored at −20°C for up to 1 year. Some reagents may freeze and should be well mixed after defrosting.)
Procedure. Four milliliters of distilled water is pipetted into a shaking tube that holds 10 to 15 mL and can be closed with either an ether-tight glass or polyethylene stopper. Then 0.2 mL of plasma is added and the tube is mixed well. The tube is placed in a boiling water bath for 4 minutes. One milliliter of 20% trichloroacetic acid is immediately added, followed by thorough mixing of the tube. The tube is centrifuged for 10 minutes at 3,000 rpm. Three milliliters of the supernatant is transferred to a test tube and 0.5 mL of 3 N HCl is added and mixed. The tubes are sealed and placed in a boiling water bath. After 1 hour, hydrolysis is complete. The tubes are allowed to cool and any condensation drops that may have formed along the walls are washed down. One-half milliliter of 0.1% sodium nitrite is added, mixed, and allowed to stand for 6 minutes. Sulfaminic acid (0.5%) is added to the tube, which is mixed by shaking until no gas bubbles are released. One-half milliliter of 0.1% aqueous α-naphthylethylenediamine dihydrochloride is added and mixed. The solution can be measured spectrophotometrically, as described later, in approximately 20 to 60 minutes.
For urine samples, the procedure is the same as that described for plasma, except that in the assay for active sulfonamides, 0.15 mL of urine is mixed with 5 mL of McIlvain buffer, and 1 mL of this mixture is added to ethyl acetate. In the assay for total sulfonamides, 0.5 mL of urine is diluted with 20 to 50 mL of distilled water (depending on the amount of sulfonamide present). Standards in a similar solvent should be prepared to cover the range of sulfonamide concentrations that are expected in the sample and run in parallel with the patient samples. In addition, distilled water and a sample of the same type of specimen (e.g., plasma or urine) from a patient who has not received sulfonamides are also run as negative controls. All tubes are assayed at 554 nm. The value for each tube is determined by subtracting from the reading for each patient sample the reading obtained from the body fluid that does not contain sulfonamides. A standard Beer’s Law or Beer-Lambert Law type of curve is constructed. One can interchange standard curves among the sulfonamides.
Occasionally, there may be so much sulfonamide present in a sample that it may not fall on the straight portion of the line. In these instances, samples should be diluted in the same body fluid from which they were collected and the assay should be repeated. If the identical body fluid is not available, distilled water can be used as a diluent with little error for all body fluids except bile. These methods are satisfactory for all sulfonamides.
Trimethoprim
Trimethoprim (TMP) can be extracted from body fluids with chloroform at a basic pH, extracted back into dilute H2SO4, and oxidized with KMnO4 in an alkaline milieu to yield the fluorescent trimethoxybenzoic acid (TMBA) (144).
Reagents. The following reagents are used:
1. 0.1 N sodium carbonate solution
2. analytical-grade chloroform
3. 0.01 N H2SO4
4. 0.1 mol/L KMnO4 in 0.1 N NaOH
5. 35% formaldehyde
6. 1 N H2SO4
Procedure. For the production of standard solutions of TMP, (a) 29.24 mg of TMBA is weighed in a 100-mL volumetric flask, which is then filled with chloroform; and (b) 5 mL of the solution from step (a) is added to a 100-mL volumetric flask, which is then filled with chloroform (this yields 20 µg/mL TMP). From this working standard solution, dilutions can be made in chloroform to cover the range of TMP that is encountered in biologic material.
To extract the antibiotic, 8 mL of Na2CO3, 10 mL of chloroform, and 1 or 2 mL of biologic fluid are added to a 25-mL shaking tube. The tube is stoppered and, to avoid emulsion, inverted gently head-over-tail for 4 minutes. The tube is centrifuged for 10 minutes at 3,000 rpm. As much of the aqueous phase as possible is aspirated into a new shaking tube and 4 mL of 0.01 N H2SO4 and 4 mL of the chloroform extract from the previous step are added. The tube is mixed by shaking for 10 minutes and centrifuged as described.
For oxidation of TMP to TMBA, 3 mL of the H2SO4 extract and 2 mL of the alkaline KMnO4 solution are added to a shaking tube. The tube is mixed and placed in a 60°C water bath for 20 minutes. Then 0.3 mL of formaldehyde is added and the tube is mixed. One milliliter of 1 N H2SO4 is added, and the tube is placed in a 60°C water bath for 20 minutes. The tube is mixed and cooled to room temperature.
For extraction of TMBA, 2 mL of chloroform is added and the tube is mixed by vigorous shaking for 10 minutes, followed by centrifugation at 3,000 rpm. The clear chloroform phase is transferred into a quartz cuvette, and fluorescence is measured at an activation wavelength of 375 nm and a fluorescence wavelength of 360 nm. The amount of TMP is determined by
C = (Ma)(Cs)(F)/Ms,
where C is the concentration of TMP in the sample, Cs is the concentration of drug in the standard, Ma is the fluorescence of the TMP-containing sample after blank subtraction, Ms is the fluorescence reading of the standard after subtraction of the chloroform bank, and F is a constant that takes into account the conversion yield of TMP in TMBA (for 1 mL of body fluid, it is 5.442 and for 2 mL, it is 2.721).
Plotting the amount of TMBA and TMP (obtained through the conversion factor) versus fluorescent intensity should yield straight lines. The method is useful for the determination of TMP in samples from all body fluids. It has a sensitivity of 20 ng of TMP/mL of plasma. Other drugs, including the sulfonamides, do not react in this procedure. The method determines the concentration of unmetabolized, active TMP.
Chloramphenicol
CAM is used for the therapy of acute bacterial meningitis, rickettsial infections, typhoid fever, and brain abscesses. However, CAM may cause hematologic toxicity. Rarely, aplastic anemia with a high fatality rate occurs. In addition, CAM can cause bone marrow suppression, which is reversible and dose-related. Toxicity occurs in relation to dose when plasma levels exceed 80 mol/L (25 µg/mL). Therapeutic monitoring of serum CAM concentrations may be helpful in evaluating and maintaining effective levels of this potentially toxic antibiotic. This may be particularly important for patients with compromised liver status. In premature neonates, metabolism of the drug is unpredictable. High serum levels in these infants can produce the fatal gray baby syndrome.
In one useful chemical method for the determination of CAM concentrations, the drug is extracted from clinical specimens in isoamyl acetate. After extraction, the CAM concentration can be determined by analysis of the yellow color that develops when the extract reacts with isonicotinic acid hydrazine and sodium hydroxide (145). It appears that many of the biologic breakdown products of CAM are not extractable by this procedure.
Duplicate standards are prepared at 10, 20, and 30 µg/mL. Two milliliters of a phosphate buffer (0.1 mol/L; 2.21 µg of NaH2PO4 · H2O and 7.61 g of Na2HPO4 · H2O in 1 L of distilled water) is added to all tubes. Then 0.5 to 1.0 mL of serum or a standard solution is added. Distilled water is added to an additional tube as a negative control. Three milliliters of isoamyl acetate (Fisher Scientific Co, Hampton, NH) is pipetted into each tube. Each tube is tightly stoppered, mixed well by shaking for approximately 10 minutes, and centrifuged (16,000 rpm in a Sorvall RC-2 centrifuge [Thermo Fischer Scientific, Inc., Waltham, MA, Norwalk, CT] is optimal). To each tube that contains 2 mL of the supernatant solvent, 1.0 mL each of 1.5 N NaOH and 3% isonicotinic acid hydrazide (Eastman Kodak Co, Rochester, NY) are added. The tubes are then stoppered and incubated in a water bath at approximately 30°C for 45 minutes. The tubes are agitated periodically to ensure good mixing. After the incubation step has been completed, the yellow underlayer is aspirated with a Pasteur pipette and the absorbance is measured with a spectrophotometer at 430 nm. The blank is read as the negative control. The standard dose-response curve is then constructed by plotting the absorbance at 430 nm (the reading obtained from the test less the reading obtained with the blank) on the x axis versus the logarithm of antibiotic concentration on the y axis. Another colorimetric assay for CAM is described next.
Reagents. The enzyme reagent includes 200 mmol/L glycylglycine, pH 8, 1 mmol/L magnesium chloride, 7 mmol/L oxamic acid, 0.09 mmol/L acetyl coenzyme A, 60 U/L CAM acetyltransferase (CAT), 90.5 mmol/L nicotinamide adenine dinucleotide (NAD), 0.2 mmol/L thiamine pyrophosphate (TPP), 0.6 mmol/L 2-oxoglutarate, 20 U/L 2-oxoglutarate dehydrogenase (2-OGDH), and 0.01% GAFAC RE-610 (GAF Corp, Wayne, NJ). This reagent must be used within 2 hours of preparation. However, if the acetyl coenzyme A is omitted, the reagent can be stored at 4°C for 24 hours without a significant loss in activity of either enzyme. The color reagent, a solution of 0.2 mmol/L 2-(2-benzothiazolyl)-5-styryl-3-(4-phthalhydrazidyl) tetrazolium chloride in 12 mmol/L citric acid, that contains 0.02 mmol/L 1-methoxy-phenazine methosulfate, 0.04% Nonidet P-40, and 0.1% sodium azide, is stored in a dark bottle at 4°C. Citric acid provides maximum reagent stability.
Procedure. The enzymatic reactions are individually optimized with respect to buffer type, pH, and substrate and cofactor concentrations. To facilitate a rapid reaction, acetyl coenzyme A is required by CAT at a concentration that is in excess of the sample CAM concentration. The 2-OGDH reaction requires the substrate 2-oxoglutarate and the cofactors NAD, TPP, and magnesium ions. Oxamic acid is included as an inhibitor of endogenous serum lactate dehydrogenase activity. When the two enzymatic reactions are combined, glycylglycine buffer (200 mmol/L, pH 8) facilitates rapid reactions and gives maximum enzyme stability.
Dehydrogenase activity is detected by using reduction of the tetrazolium salt 2-(2-benzothiazolyl)-5-styryl-3-(4-phthalhydrazidyl) tetrazolium chloride (146) to a formazan dye, with the highest molar extinction coefficient under the prevailing assay conditions. CAT activity is determined by measuring the increase in absorbance at 412 nm of an assay mixture containing 100 mmol/L Tris-HCl, pH 8.0, 0.1 mmol/L acetyl coenzyme A, 0.1 mmol/L CAM, and 1 mmol/L 5,5-dithiobis (2-nitrobenzoic acid) (Ellman’s reagent or DTNB). The reaction is initiated by adding 25 µL of CAT to 1 mL of assay mixture in a semimicrocuvette (path length of 1 cm).
2-OGDH (EC 1.2.4.2) activity is determined by measuring the increase in absorbance at 340 nm at 30°C of an assay mixture (1 mL) that contains 50 mmol/L of potassium phosphate buffer, pH 8.0, 1 mmol/L MgCl2, 2.5 mmol/L NAD, 0.2 mmol/L TPP, 0.1 mmol/L coenzyme A, 2.5 mmol/L cysteine, and 2 mmol/L 2-oxoglutarate. The reaction is initiated with 25 µL of 2-OGDH.
The serum sample or CAM standard (0.1 mmol/L, 32 mg/L, 0.1 mL) is added to the enzyme reagent (0.5 mL) in a semimicrocuvette (path length of 1 cm), mixed well, and incubated at room temperature for 4 minutes. Color reagent (0.5 mL) is added and, after incubation at room temperature for exactly 2 minutes, the absorbance of the reaction mixture is measured at 575 nm. After 2 minutes, the reaction mixture exhibits a gradual increase in absorbance. A sample blank is prepared and its absorbance is measured by following the same procedure, except that glycylglycine buffer (200 mmol/L, pH 8) is substituted for the enzyme reagent.
The method is based on the reduction of a pale tetrazolium salt to a strongly colored formazan dye by NADH. The thiol groups of 2-OGDH also act as reducing agents, causing formazan production independent of the NADH reaction. Therefore, a reagent blank is required in addition to the sample blank, the absorbance of which is added to the sample blank value. The absorbance of the reagent blank is constant.
Performance. The assay is linear over the CAM range of 5 to 200 mol/L. The intra- and interbatch coefficients of variation (precision) are 1.4% to 4.9% and 4.3% to 6.3%, respectively. Mean recoveries (accuracy) from CAM-spiked (0.1 mmol/L and 0.025 mmol/L) serum samples from normal individuals and patients with renal failure were 98.4% and 105.6% for serum from normal individuals and 100.9% and 106.8% for serum from patients with renal failure. The method does not detect the inactive prodrug forms of CAM, that is, CAM succinate (intravenous preparations) and CAM palmitate (oral suspensions). Of the metabolites tested (CAM base, reduced base, and glycolic acid), only glycolic acid is recognized by CAT, with a cross-reactivity of 81%. However, this is a minor metabolite (<3%) and its detection is not considered important to the clinical utility of the assay.
Previous work indicates that CAT does not recognize CAM glucuronide (147). The method correlates well with reversed-phase high-performance liquid chromatography (RP-HPLC), which is specific for microbiologically active CAM. The assay also detects thiamphenicol, a CAM analog, with similar sensitivity and with a linear response up to a serum concentration of 100 µM. The endogenous colored compounds bilirubin and hemoglobin interfere with the color reaction when they are present in serum at concentrations above 200 µM and 0.2 mg/dL, respectively, because of a shift in the optimum wavelength of the final color. Thus, because of colorimetric interference, this assay is not recommended for use with grossly hemolyzed samples or when bilirubin concentrations exceed 200 µM.
A main advantage of this procedure is the speed with which an accurate CAM measurement can be obtained. The method requires no pretreatment of the sample such as heating or solvent extractions, as is required for HPLC, the more commonly employed assay for CAM. Moreover, a result is available within 6 minutes. The precision of the assay described here, when performed manually, is similar to that of the automated enzyme-multiplied immunoassay technique (EMIT) (148). However, it has the advantages of producing a linear response (allowing a single-point calibration) and requiring only a simple spectrophotometer to measure absorbance. Because it is a two-reagent system, the assay may also be adapted for a wide range of discrete analyzers. The method is specific and shows no significant interference by high concentrations of urea, creatinine, or phenolic compounds, which may be present in the serum of patients with renal failure (147).
RADIOENZYME ASSAYS
Bacteria are often resistant to antibiotics because they produce inactivating enzymes. Benveniste and Davies (149) and Davies et al. (150) provided the basis for radioenzymatic techniques in their description of a method by which they could determine the types of enzymes that inactivated certain antibiotics. Their basic method is used for the theoretical study of bacterial resistance.
Radioenzyme assays have been largely replaced. They were originally a by-product of the study of how enzymes destroy aminoglycosides and CAM. Table 8.2 presents the general advantages and disadvantages of radioenzymatic assays. Radioactive ATP in the presence of adenylating enzyme transfers radioactive 14C to the aminoglycoside. Aminoglycosides, which are positively charged, stick to negatively charged phosphocellulose papers. By enumerating the radioactive counts on these phosphocellulose papers, the extent to which adenylation occurred can be measured. Acetylating enzymes, which transfer acetyl groups from radioactive acetyl coenzyme A to aminoglycosides, can be similarly employed, with the measured counts on phosphocellulose paper reflecting the amount of acetylation that took place (149,150).
These adenylation and acetylation reactions provide the basis for radioenzymatic assays. It should be noted that the reactions are stoichiometric in that the amount of transfer in both adenylation and acetylation reactions is directly related to the quantity of antibiotic in the solution. The reaction takes place in several steps.
Many authors have used a method by which periplasmic enzymes are released from bacteria due to changes in osmotic pressure (151,152). By this method, less than 4% of the intracellular bacterial contents are released. However, the method is time-consuming and technically involved. Sonication has been investigated as a faster and easier method of obtaining the enzyme (153). The enzymes obtained by sonication appear to be as effective as those produced by osmotic shock. However, enzymes obtained by sonication may be somewhat more contaminated and unstable because of the release of proteolytic enzymes inside the bacteria (154). This could limit the length of time that the enzyme can be stored, a critical factor in the long-term usage of radioenzymatic techniques (155).
One may prepare the sonicate as follows. A 16-hour culture of E. coli RS/W677 is centrifuged at 14,000 × g for 5 minutes. The sediment is washed twice in 30 mmol/L NaCl plus 10 mmol/L Tris-HCl, pH 7.8. After the second wash, the pellet is suspended in 0.5 mmol/L MgCl2 at 4°C (5 mL of 0.5 mmol/L MgCl2 per 100 mL of original culture volume). The suspension is then sonicated for 20 seconds using a Dawe-type 3057A Soniprobe (Dawe Instruments Ltd, London, United Kingdom) set to give a 4 amp current. The cellular debris is removed by centrifugation at 25,000 × g for 20 minutes. The supernatant is divided into 0.1-mL aliquots and stored at 20°C (94).
Because it is undesirable to have to prepare the enzyme frequently, its stability has been studied under various conditions (154). Several methods have been developed to decrease enzyme lability. Keeping the enzyme frozen and in an ice bath are effective techniques (36,156). The enzyme has been shown to be stable for 24 hours at 4°C and 30 days at −20°C with BSA (157). Storing partially purified enzyme in reducing agents appears to increase its life span (158). Freezing the enzyme at low temperatures, such as −70°C, in quantities that will be used in a day’s run, appears to be the most efficient and effective technique (159,160). It increases the storage life of the enzyme and significantly decreases the amount of technician time that is required to set up the assays.
DIRECT FLUORESCENT CHEMICAL ANALYSIS
A molecule is fluorescent when it can receive light at one wavelength and emit it at another wavelength. The wavelengths at which a given molecule receives and emits light are often specific to the molecule, a reactive group on a molecule, or a class of molecules. One major disadvantage of these assays is that other material present in the specimen, particularly radiologic fluorescein dyes and certain proteins, can fluoresce and interfere with the test. However, fluorescent assays typically are much more sensitive than chemical assays and can quantify compounds in the nanogram and often picogram per milliliter range.
Fluorescence can be determined in one of two ways. First, one may use the inherent fluorescent properties of the molecule. Second, for antibiotics that are either weakly fluorescent or nonfluorescent, one may covalently link a strongly fluorescent moiety to the drug in question. Because of the need for an extraction step, the possible need for coupling steps, the need for rather specialized equipment, the lack of standardized techniques, and the frequent inability to distinguish between active and inactive antibiotics, fluorescence methods have not been widely used in clinical laboratories, although they have been used in industry and U.S. Public Health Service laboratories. The reader is referred to the book by Undenfriend (161) for a general view of fluorescent analysis.
Although penicillins and cephalosporins are not generally inherently fluorescent, many produce fluorescent compounds under hydrolysis in the presence of acid (162). Attempts to simplify the extraction procedures and the number of technical manipulations that are required to generate such reactions have led to methods for measurement that can be clinically useful (141,163). For example, ampicillin concentrations can be determined in the absence of ampicillinoic acid by extraction. Standards should be prepared to cover the expected range of concentrations and serum blanks should be run to obtain measurements of background fluorescence.
Tetracyclines
Different tetracyclines require different fluorescent reagents to enhance their light-emitting properties (164). Each tetracycline should be tested individually to optimize these assays. Hall (165) has expanded the technique to measure the concentrations of tetracycline mixtures in plasma by using acid hydrolysis or alkaline degradation to convert the tetracycline into a fluorescent form. In these methods, aluminum salts are used to enhance the fluorescence of the end products. This fluorescence is measured with a spectrofluorometer. Each tetracycline exhibits a different structural arrangement of the chemical groups that surround the fluorescent nucleus of the anhydrous salts, creating individual fluorescent characteristics (165). A general method for the fluorometric determination of tetracyclines follows (166).
Apparatus
An Aminco-Bowman spectrophotofluorometer (American Instrument Company, Silver Spring, MD) fitted with a xenon arc lamp and an R 136 photomultiplier, or its equivalent, should be employed. Mirrors and 1-mm slits are placed in the cell housing.
Reagents
For most tetracyclines, one can use 0.5 mol/L magnesium acetate tetrahydrate plus 0.3 mol/L sodium barbitone in ethanedial. Minocycline, and other 7-aminotetracyclines, can be measured with a mixture of 0.2 mol/L magnesium acetate and 0.2 mol/L citric acid in ethanedial.
Procedure
A 0.2-mL aliquot of the sample (serum or other fluid) is mixed with 0.4 mL of a phosphate buffer (3 mol/L NaH2PO4 plus 1 mol/L Na2SO3) and thoroughly extracted with 2.5 mL of amyl acetate. The phases are separated by allowing them to settle or centrifuging the tube (approximately 500 × g). Two milliliters of the organic (top) phase are transferred to a Brown fluorometer cuvette. A suitable fluorescence reagent (0.6 mL) is added. (Fluorescence reagents are described later.) The tubes are mixed by shaking for 5 minutes. The turquoise fluorescence in the lower phase is read 20 minutes or more after shaking. If the lower phase is cloudy, the tubes are centrifuged (500 × g for 2 or 3 minutes) before reading. Standards (0.2 mL of a 10 mmol/L solution of the appropriate tetracycline) and blanks (0.2 mL of water) are also made.
For the determination of minocycline, a pH 6.5 buffer (0.5 mol/L NaH2PO4 plus 0.5 mol/L Na2HPO4) should be used instead of the phosphate sulfite buffer (137). Fluorescence is measured with excitation at 405 nm and emission at 490 nm. The fluorescence of 7-aminotetracyclines is read with excitation at 380 nm and emission at 480 nm (166).
IMMUNOLOGIC ASSAYS
Immunologic assays came into use in the mid-1970s. These assays were based largely on existing equipment and merely exploited procedures available for the assay of hormones. However, for the first time, the assay of antibiotics was removed from the realm of the specialist. These assays could be performed in a central location, with the instrument playing the primary role. The development of the means to elicit specific, high-titered antibodies to hapten antibiotics allowed RIA techniques to deliver a specificity that was impossible with biologic agents. As with any nonbiologic assay, however, one always had to be particularly careful not to measure a nonactive metabolite.
Because RIA equipment was expensive, the measurement of antibiotics was often mixed in with the assay of other drugs, resulting in significant delays in processing. The requirement to maintain a stock of highly active radioisotopes (often with short half-lives) gave impetus to the development of nonisotopic immunoassays. Unlike hormones, which are present in extremely small amounts and require highly sensitive methods of detection, antibiotics are generally present in levels above 0.5 µg/mL. Because other small molecules, such as antiepileptics and drugs of abuse, are also present in these levels, a technology was developed to measure small molecules by somewhat less sensitive, nonisotopic means. Unlike the stimulatory role played by existing RIA equipment in the development of RIA, the impetus for the development of the new nonisotopic immunoassays was largely the need to assay the aminoglycoside class of antibiotics.
Before the application of this technique to the measurement of antibiotic concentrations in 1975, RIA had been used for a number of years for the quantitation of hormonal substances (167). All RIAs of antibiotics employ three broad reaction steps. The first step involves three components: radiolabeled antigen (antibiotic); high-titer, high-avidity antibody to that antigen; and unlabeled antigen (antibiotic obtained from the patient’s serum). The reaction with either labeled or unlabeled antigen produces antibody combined with labeled antigen or antibody combined with unlabeled antigen. The more unlabeled antigen that is present in the reaction mixture, the less radiolabeled antigen combines with antibody.
After equilibrium is reached, one must quantify the amount of bound antibody in the mixture. This step involves either the removal of the bound antigen-antibody complex from solution by a precipitating agent or the removal of the unbound antigen from solution by chemical means. Because antigen/antibody reactions are stoichiometric in nature, the quantification of either the bound radiolabeled or the free radiolabeled antigen is directly proportional to the antibiotic level in a sample (168–170).
Although there are individual modifications, the test procedures follow a basic course (167). First, one incubates a known quantity of tritiated antibiotic with antibody (of known potency) to that antibiotic. A given amount of patient’s serum is added and the mixture is allowed to come to equilibrium. These assays are heterogeneous and produce sigmoidal coprecipitation curves. Early investigators were hampered because only the relatively linear part of the sigmoidal curve could be used for the calculation of antibiotic concentrations. Robard et al. (171) devised a method in which the sigmoidal curve was converted to a straight line, thereby permitting the calculation of results over a much wider range of values. They found that the plot of y = 100 (B/B°) may be linearized by the equation representing a straight line in which logit (y) = A + B log x, where x is the amount of unlabeled or patient drug, and y represents the percentage of antibiotic that is bound.
This logit conversion not only allowed a much wider range of antibiotic concentrations to be measured but also permitted the development of standard curves. As a result, one did not have to repeat all the controls each time a specimen was run. An advantage of immunologic assays is their within-class specificity. Although there may be cross-reactivity among members of the same drug class, there is no cross-reactivity among members of different classes (e.g., gentamicin and vancomycin).
Nonisotopic Immunoassays
The development of EIAs by Engvall and Perlmann (172,173) laid the foundation for design of nonradioisotopic immunoassays. In these immunoassays, an enzyme or fluorometric substance is coupled to the antigen or antibody in such a way that the activity of the parent compound is not appreciably affected. The significant difference between nonisotopic immunoassays and RIAs is the means of counting the label. In nonisotopic assays, the quantity label present is estimated from enzyme or fluorescence activity, which changes under the assay conditions.
Generally, when an enzyme–substrate reaction is used in an immunoassay, enzyme activity is measured as reaction velocity. In this situation, the reaction velocity must be proportional to the number of enzyme molecules that catalyze the reaction (174). In fluorescence immunoassays, a change in the nature of the interaction of light with the substrate is measured (175).
In many nonisotopic immunoassays, the label, whether fluorescent or nonfluorescent, is an enzyme. In order to distinguish RIAs from nonisotopic immunoassays, we must consider the characteristics of an enzyme label. First, it must be recognized easily as the label. The enzyme must be attached to the substrate in such a way that it maintains activity yet performs satisfactorily under the assay conditions. Second, the label must be stable. The label must not disintegrate when the assay is in process and it must be stable during storage. Invariably, the enzyme is covalently bound to the labeled molecule. Third, the enzyme must be quantitatively measurable. In nonisotopic immunoassays, one measures a secondary reaction product (i.e., produced by a substrate acting on the enzyme), rather than direct release of a radioisotope (176,177). Therefore, we do not measure the enzyme molecules themselves but instead measure the catalyzed reaction processes. With any enzymatic label, the product that is produced must be proportional to the amount of enzyme that catalyzes the reaction.
In considering the theory of enzyme activity and its relationship to the clinical assay, we must remember the Michaelis-Menten equation:
v = V [S/(S + Km)],
where v is the measured reaction velocity, S is the concentration of substrate, and Km is the calculated constant. In practice, one attempts to design the assay conditions so that S is much greater than Km. In this case, we may assume that v is approximately equal to V and that
v = (k)E2,
where E2 is the total amount of enzyme that is present in the reaction mixture. It should be noted that this equation implies that the reaction velocity is independent of time.
The equations just described require that the reactions be followed continuously as they occur. One must measure the accumulation of product or the change in fluorescence as it occurs over time. There have been three primary means of quantitatively following enzyme reactions for the assay of haptens, including antibiotics, under these conditions. First is the spectrophotometric assay. The spectrophotometer is useful when the product of the reaction can be measured between 190 and 800 nm. Most workers have attempted to measure the release of the product directly. However, the product may first have to be transformed into a colored product. The advantage of using a spectrophotometer is that this instrument is readily available in clinical laboratories.
Much work has involved the study of fluorometric methods, which is the second approach. The outstanding feature of fluorometry is its high degree of sensitivity. Fluorometic methods are generally 100 to 1,000 times more sensitive than spectrophotometric methods. This sensitivity is especially manifested either when the initial substrate is nonfluorescent and becomes so during the reaction or when there is a change in fluorescence polarization as the reaction proceeds.
The third method, which employs electrodes, offers several advantages. Hydrogen ion concentrations and automatic titration apparatus may be employed to continuously measure a reaction as it proceeds. Considerable work has been performed on an oxygen electrode, which is a type of polarography (54).
Fundamental to nonisotopic immunoassays is the preparation of hapten–protein conjugates, in which haptens (antibiotic) and proteins (generally an enzyme) both function naturally. Most of the described methods have attached the hapten to the protein by free amino, hydroxyl, or carboxyl groups. Haptens with hydroxyl groups may be conjugated to a carrier protein by activation of the carboxyl group, followed by acylation of amino groups in the protein. The mixed-anhydride procedure has been commonly used for this purpose. Figure 8.6 demonstrates the principles of the reaction. The procedure is performed directly with the hapten and the conjugate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


