35
Antiviral Drugs
CHAPTER CONTENTS
PRINCIPLES OF ANTIVIRAL THERAPY
Compared with the number of drugs available to treat bacterial infections, the number of antiviral drugs is very small. The major reason for this difference is the difficulty in obtaining selective toxicity against viruses; their replication is intimately involved with the normal synthetic processes of the cell. Despite the difficulty, several virus-specific replication steps have been identified that are the site of action of effective antiviral drugs (Table 35–1). Table 35–2 describes the mode of action of antiviral drugs that block early events in viral replication, and Table 35–3 describes the mode of action of antiviral drugs that block viral nucleic acid synthesis. Figure 35–1 shows the replication of a model virus and the site of action of drugs used to treat various viral infections. Figure 35–2 shows the replication of human immunodeficiency virus (HIV) and the site of action of drugs used to treat HIV infection.
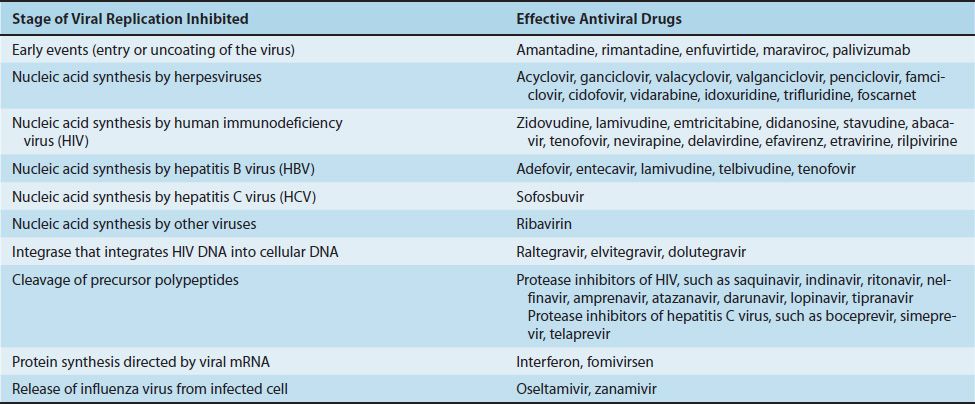
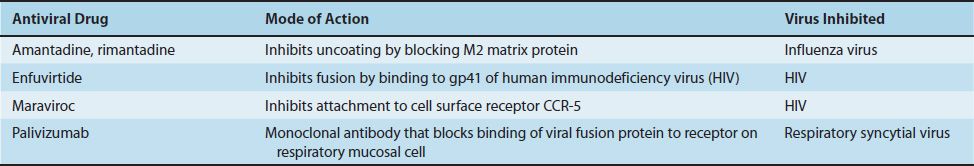
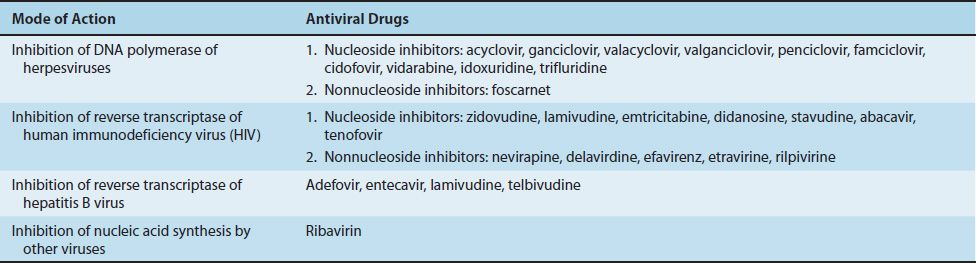
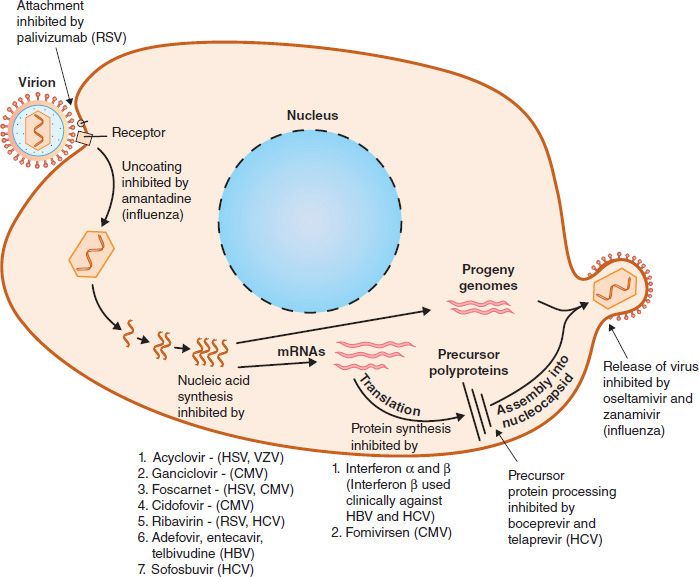
FIGURE 35–1 Replicative cycle of a model virus showing the site of action of drugs used to treat various viral infections. CMV, cytomegalovirus; HBV, hepatitis B virus; HCV, hepatitis C virus; HSV, herpes simplex virus; RSV, respiratory syncytial virus; VZV, varicella-zoster virus.
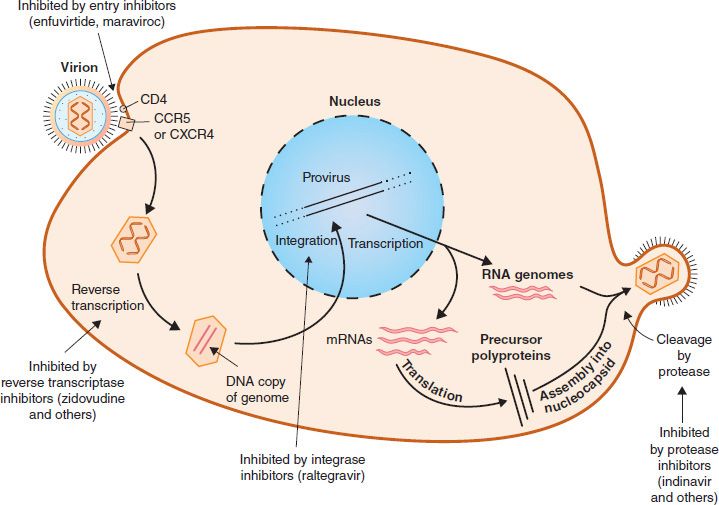
FIGURE 35–2 Replicative cycle of human immunodeficiency virus (HIV) showing the site of action of drugs used to treat HIV infection.
Another limitation of antiviral drugs is that they are relatively ineffective because many cycles of viral replication occur during the incubation period when the patient is well. By the time the patient has a recognizable systemic viral disease, the virus has spread throughout the body and it is too late to interdict it. Furthermore, some viruses (e.g., herpesviruses) become latent within cells, and no current antiviral drug can eradicate them.
Another limiting factor is the emergence of drug-resistant viral mutants. For example, when drug-resistant mutants of HIV emerge, it requires that drug regimens be changed. Also, treatment of HIV infection uses multiple drugs, often from different classes, so that if mutants resistant to one drug emerge, another drug will still be effective.
INHIBITION OF EARLY EVENTS
Amantadine (a-adamantanamine, Symmetrel) is a three-ring compound (Figure 35–3) that blocks the replication of influenza A virus. It prevents replication by inhibiting uncoating of the virus by blocking the “ion channel” activity of the matrix protein (M2 protein) in the virion. Absorption and penetration occur normally, but transcription by the virion RNA polymerase does not because uncoating cannot occur. This drug specifically inhibits influenza A virus; influenza B and C viruses are not affected.
Despite its efficacy in preventing influenza, it is not widely used in the United States because the vaccine is preferred for the high-risk population. Furthermore, most isolates have become resistant to amantadine. The main side effects of amantadine are central nervous system alterations such as dizziness, ataxia, and insomnia. Rimantadine (Flumadine) is a derivative of amantadine and has the same mode of action but fewer side effects.
Enfuvirtide (Fuzeon) is a synthetic peptide that binds to gp41 on the surface of HIV, thereby blocking the entry of the virus into the cell. It is the first of a new class of anti-HIV drugs known as “fusion inhibitors” (i.e., they prevent the fusion of the viral envelope with the cell membrane).
Maraviroc (Selzentry) blocks the binding of HIV to CCR-5—an important coreceptor for those strains of HIV that use CCR-5 for entry into the cell. The drug binds to CCR-5 and blocks the interaction of gp120, an HIV envelope protein, to CCR-5 on the cell surface.
Palivizumab (Synagis) is a monoclonal antibody directed against the fusion protein of respiratory syncytial virus (RSV). Palivizumab neutralizes RSV by binding to the fusion protein on the surface of RSV, thereby preventing the virus from binding to receptors on the surface of respiratory tract mucosal cells. It is used to prevent bronchiolitis and pneumonia in premature or immunocompromised infants.
INHIBITION OF VIRAL NUCLEIC ACID SYNTHESIS
Inhibitors of Herpesviruses
Nucleoside Inhibitors
These drugs are analogues of nucleosides that inhibit the DNA polymerase of one or more members of the herpesvirus family. For example, acyclovir inhibits the DNA polymerase herpes simplex virus types 1 and 2 (HSV-1 and -2) and varicella-zoster virus but not cytomegalovirus (CMV).
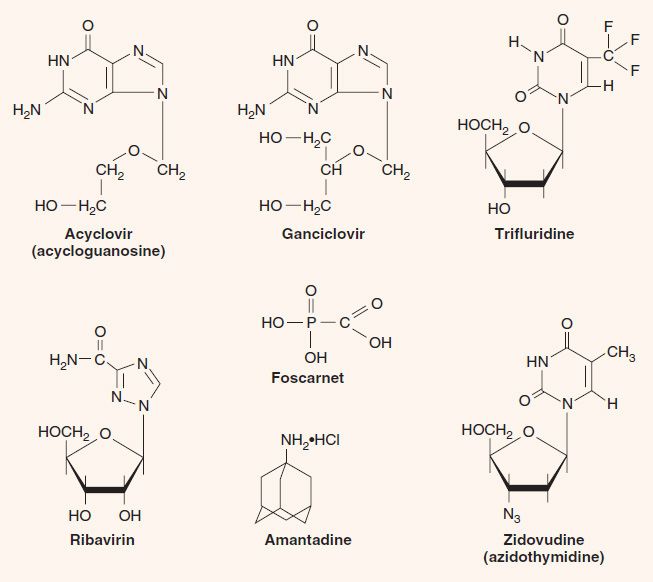
1. Acyclovir—Acyclovir (acycloguanosine, Zovirax) is a guanosine analogue that has a three-carbon fragment in place of the normal sugar, ribose, which has five carbons (see Figure 35–3). The term acyclo refers to the fact that the three-carbon fragment does not have a sugar ring structure (a = without, cyclo = ring).
Acyclovir is active primarily against HSV-1 and -2 and varicella-zoster virus (VZV). It is relatively nontoxic, because it is activated preferentially within virus-infected cells. This is due to the virus-encoded thymidine kinase, which phosphorylates acyclovir much more effectively than does the cellular thymidine kinase. Because only HSV-1, HSV-2, and VZV encode a kinase that efficiently phosphorylates acyclovir, the drug is active primarily against these viruses. It has no activity against CMV. Once the drug is phosphorylated to acyclovir monophosphate by the viral thymidine kinase, cellular kinases synthesize acyclovir triphosphate, which inhibits viral DNA polymerase much more effectively than it inhibits cellular DNA polymerase. Acyclovir causes chain termination because it lacks a hydroxyl group in the 3´ position.
To recap, the selective action of acyclovir is based on two features of the drug. (1) Acyclovir is phosphorylated to acyclovir monophosphate much more effectively by herpesvirus-encoded thymidine kinase than by cellular thymidine kinase. It is therefore preferentially activated in herpesvirus-infected cells and much less so in uninfected cells, which accounts for its relatively few side effects. (2) Acyclovir triphosphate inhibits herpesvirus-encoded DNA polymerase much more effectively than it does cellular DNA polymerase. It therefore inhibits viral DNA synthesis to a much greater extent than cellular DNA synthesis (Figure 35–4).
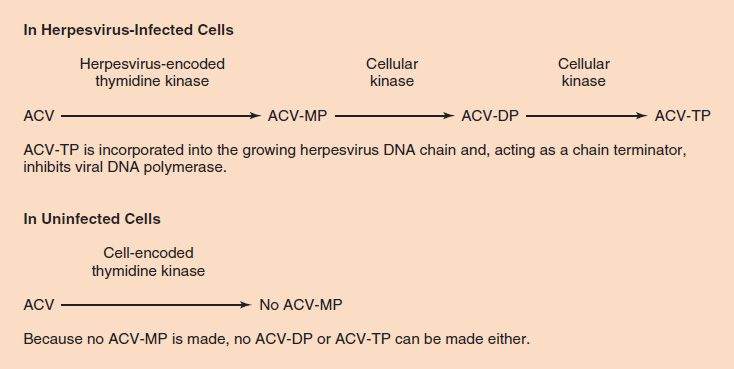
FIGURE 35–4 Acyclovir (ACV) is phosphorylated to ACV-MP very effectively by herpesvirus-encoded thymidine kinase but very poorly by cell-encoded thymidine kinase. The thymidine kinases encoded by herpes simplex virus (HSV)-1, HSV-2, and varicella-zoster virus (VZV) are particularly active on ACV; the thymidine kinases encoded by cytomegalovirus and Epstein–Barr virus are not. This accounts for the selective action of ACV in cells infected by HSV-1, HSV-2, and VZV. The fact that ACV-TP is not made in uninfected cells explains why ACV has so few side effects (i.e., why DNA synthesis is not inhibited in uninfected cells). ACV-MP, ACV monophosphate; ACV-DP, ACV diphosphate; ACV-TP, ACV triphosphate.
Topical acyclovir is effective in the treatment of primary genital herpes and reduces the frequency of recurrences while it is being taken. However, it has no effect on latency or on the rate of recurrences after treatment is stopped. Acyclovir is the treatment of choice for HSV-1 encephalitis and is effective in preventing systemic infection by HSV-1 or VZV in immunocompromised patients.
Acyclovir-resistant mutants have been isolated from HSV-1- and VZV-infected patients. Resistance is most often due to mutations in the gene encoding the viral thymidine kinase. This results in reduced activity of or the total absence of the virus-encoded thymidine kinase.
Acyclovir is well tolerated and causes few side effects—even in patients who have taken it orally for many years to suppress genital herpes. Intravenous acyclovir may cause renal or central nervous system toxicity.
Derivatives of acyclovir with various properties are now available. Valacyclovir (Valtrex) achieves a high plasma concentration when taken orally and is used in herpes genitalis and in herpes zoster. Penciclovir cream (Denavir) is used in the treatment of recurrent orolabial herpes simplex. Famciclovir (Famvir), when taken orally, is converted to penciclovir and is used to treat herpes zoster and herpes simplex infections.
2. Ganciclovir—Ganciclovir (dihydroxypropoxymethylguanine, DHPG, Cytovene) is a nucleoside analogue of guanosine with a four-carbon fragment in place of the normal sugar, ribose (see Figure 35–3). It is structurally similar to acyclovir but is more active against CMV than is acyclovir. Ganciclovir is activated by a CMV-encoded phosphokinase in a process similar to that by which acyclovir is activated by HSV. Isolates of CMV resistant to ganciclovir have emerged, mostly due to mutations in the UL97 gene that encodes the phosphokinase.
Ganciclovir is effective in the treatment of retinitis caused by CMV in patients with acquired immunodeficiency syndrome (AIDS) and is useful in other disseminated infections, such as colitis and esophagitis, caused by this virus. The main side effects of ganciclovir are leukopenia and thrombocytopenia as a result of bone marrow suppression. Valganciclovir, which can be taken orally, is also effective against CMV retinitis.
3. Cidofovir—Cidofovir (hydroxyphosphonylmethoxy-propylcytosine, HPMPC, Vistide) is an analogue of cytosine that lacks a ribose ring. It is useful in the treatment of retinitis caused by CMV and in severe human papillomavirus infections. It may be useful in the treatment of severe molluscum contagiosum in immunocompromised patients. Kidney damage is the main side effect.
4. Vidarabine—Vidarabine (adenine arabinoside, ara-A) is a nucleoside analogue with arabinose in place of the normal sugar, ribose. On entering the cell, the drug is phosphorylated by cellular kinases to the triphosphate, which inhibits the herpesvirus-encoded DNA polymerase more effectively than the cellular DNA polymerase. Vidarabine is effective against HSV-1 infections such as encephalitis and keratitis but is less effective and more toxic than acyclovir.
5. Idoxuridine—Idoxuridine (iododeoxyuridine, IDU, IUDR) is a nucleoside analogue in which the methyl group of thymidine is replaced by an iodine atom. The drug is phosphorylated to the triphosphate by cellular kinases and incorporated into DNA. Because IDU has a high frequency of mismatched pairing to guanine, it causes the formation of faulty progeny DNA and mRNA. However, because IDU is incorporated into normal cell DNA as well as viral DNA, it is too toxic to be used systemically. It is clinically useful in the topical treatment of keratoconjunctivitis caused by herpes simplex virus, but in the United States, trifluorothymidine (see next entry) is the drug of choice.
6. Trifluridine—(trifluorothymidine, Viroptic) is a nucleoside analogue in which the methyl group of thymidine contains three fluorine atoms instead of three hydrogen atoms (see Figure 35–3). Its mechanism of action is the same as that of IDU. Like IDU, it is too toxic for systemic use. It is the drug of choice for the topical treatment of keratoconjunctivitis caused by herpes simplex virus.
Nonnucleoside Inhibitors
Nonnucleoside inhibitors inhibit the DNA polymerase of herpesviruses by mechanisms distinct from the nucleoside analogues described previously. Foscarnet is the only approved drug in this class at this time.
1. Foscarnet—Foscarnet (trisodium phosphonoformate, Foscavir), unlike the previous drugs, which are nucleoside analogues, is a pyrophosphate analogue (see Figure 35–3). It binds to DNA polymerase at the pyrophosphate cleavage site and prevents removal of the phosphates from nucleoside triphosphates (dNTP). This inhibits the addition of the next dNTP and, as a consequence, the extension of the DNA strand. Foscarnet inhibits the DNA polymerases of all herpesviruses, especially HSV and CMV. Unlike acyclovir, it does not require activation by thymidine kinase. It is useful in the treatment of retinitis caused by CMV, but ganciclovir is the treatment of first choice for this disease. Foscarnet is also used to treat patients infected with acyclovir-resistant mutants of HSV-1 and VZV.
Inhibitors of Retroviruses
Nucleoside Inhibitors
The selective toxicity of zidovudine, lamivudine, emtricitabine, didanosine, zalcitabine, stavudine, abacavir, and tenofovir is based on their ability to inhibit DNA synthesis by the reverse transcriptase of HIV to a much greater extent than they inhibit DNA synthesis by the DNA polymerase in human cells. These drugs are collectively called nucleoside reverse transcriptase inhibitors (NRTIs). The effect of these drugs on the replication of HIV is depicted in Figure 35–2.
1. Zidovudine—Zidovudine (azidothymidine, Retrovir, AZT) is a nucleoside analogue that causes chain termination during DNA synthesis; it has an azido group in place of the hydroxyl group on the ribose (see Figure 35–3). It is particularly effective against DNA synthesis by the reverse transcriptase of HIV and inhibits the growth of the virus in cell culture. The main adverse effects of zidovudine are bone marrow suppression and myopathy.
2. Lamivudine—Lamivudine (dideoxythiacytidine, Epivir, 3TC) is a nucleoside analogue that causes chain termination during DNA synthesis by the reverse transcriptase of HIV. When used in combination with AZT, it is very effective both in reducing the viral load and in elevating the CD4 cell count. Lamivudine is also used in the treatment of chronic hepatitis B because it inhibits the reverse transcriptase of hepatitis B virus. It is one of the best tolerated of the nucleoside inhibitors, but adverse effects such as neutropenia, pancreatitis, and peripheral neuropathy do occur.
3. Emtricitabine—Emtricitabine (Emtriva), a derivative of lamivudine, is also useful and well tolerated. A combination of emtricitabine and tenofovir (Truvada) can be used for preexposure prophylaxis for men who have sex with men as well as for postexposure prophylaxis.
4. Didanosine—Didanosine (dideoxyinosine, Videx, ddI) is a nucleoside analogue that causes chain termination during DNA synthesis; it is missing hydroxyl groups on the ribose. The administered drug ddI is metabolized to ddATP, which is the active compound. It is effective against DNA synthesis by the reverse transcriptase of HIV and is used to treat patients with AIDS who are intolerant of or resistant to zidovudine. The main adverse effects of didanosine are pancreatitis and peripheral neuropathy.
5. Stavudine—Stavudine (didehydrodideoxythymidine, d4T, Zerit) is a nucleoside analogue that causes chain termination during DNA synthesis. It inhibits DNA synthesis by the reverse transcriptase of HIV and is used to treat patients with advanced AIDS who are intolerant of or resistant to other approved therapies. The main adverse effect is peripheral neuropathy.
6. Abacavir—Abacavir (Ziagen) is a nucleoside analogue of guanosine that causes chain termination during DNA synthesis. It is available through the “expanded access” program to those who have failed currently available drug regimens. Abacavir is used in combination with either a protease inhibitor, typically darunavir and ritonavir, or zidovudine plus lamivudine. The main adverse effects are liver damage and severe hypersensitivity reactions. Patients who have an HLA-B1701 allele are more likely to have a severe hypersensitivity reaction, such as fever, rash, or respiratory problems, to abacavir. Patients should be tested for this gene before being prescribed abacavir. If patients develop hypersensitivity symptoms, abacavir should be immediately and permanently discontinued.
7. Tenofovir—Tenofovir (Viread) is an acyclic phosphonate that is an analogue of adenosine monophosphate. It is a reverse transcriptase inhibitor that acts by chain termination. It is approved for use in patients who have developed resistance to other reverse transcriptase inhibitors and in those who are starting treatment for the first time. It should be used in combination with other anti-HIV drugs. The main adverse effects are liver damage, lactic acidosis, and renal failure.
Nonnucleoside Inhibitors
Unlike the drugs described earlier, the drugs in this group are not nucleoside analogues and do not cause chain termination. The nonnucleoside reverse transcriptase inhibitors (NNRTIs) act by binding near the active site of the reverse transcriptase and inducing a conformational change that inhibits the synthesis of viral DNA. NNRTIs should not be used as monotherapy because resistant mutants emerge rapidly. Strains of HIV resistant to one NNRTI are usually resistant to others as well. NNRTIs are typically used in combination with one or two nucleoside analogues.
1. Nevirapine—Nevirapine (Viramune) is usually used in combination with zidovudine and didanosine. There is no cross-resistance with the nucleoside inhibitors of reverse transcriptase described previously. The main side effect of nevirapine is a severe skin rash (Stevens-Johnson syndrome).
2. Delavirdine—Delavirdine (Rescriptor) is effective in combination with either zidovudine or zidovudine plus didanosine. The main side effect of delavirdine is a skin rash.
3. Efavirenz—Efavirenz (Sustiva) is effective in combination with zidovudine plus lamivudine. The most common side effects are referable to the central nervous system, such as dizziness, insomnia, and headaches.
4. Etravirine—Etravirine (Intelence) is a second-generation NNRTI useful in treatment-experienced patients who have significant viremia. It is most effective when given in combination with two protease inhibitors, darunavir and ritonavir. The most common adverse effect is a rash, and Stevens-Johnson syndrome has occurred, albeit rarely.
5. Rilpivirine—Rilpivirine (Edurant) is a second-generation NNRTI useful in treatment-naïve adult patients. It is most effective when used in combination with either tenofovir or emtricitabine. The most common adverse effects are depression and insomnia.
Inhibitors of Hepatitis B Virus
Adefovir
Adefovir (Hepsera) is a nucleotide analogue of adenosine monophosphate that inhibits the DNA polymerase (reverse transcriptase) of hepatitis B virus (HBV). It is used for the treatment of chronic active hepatitis caused by this virus.
Entecavir
Entecavir (Baraclude) is a guanosine analogue that inhibits the DNA polymerase (reverse transcriptase) of HBV. It has no activity against the DNA polymerase (reverse transcriptase) of HIV. It is approved for the treatment of adults with chronic HBV infection.
Lamivudine
Lamivudine is described in the section “Inhibitors of Retroviruses.”
Telbivudine
Telbivudine (Tyzeka) is a thymidine analogue that inhibits the DNA polymerase (reverse transcriptase) of HBV but has no effect on the reverse transcriptase of HIV. It is useful in the treatment of chronic HBV infection.
Tenofovir
Tenofovir is described in the section “Inhibitors of Retroviruses.”
Inhibitors of Hepatitis C Virus
Sofosbuvir (Sovaldi) is a uridine analogue that inhibits the RNA polymerase of HCV. It is useful in the treatment of chronic HCV infection caused by genotypes 1, 2, 3, and 4.
Inhibitors of Other Viruses
Ribavirin
Ribavirin (Virazole) is a nucleoside analogue in which a triazole-carboxamide moiety is substituted in place of the normal purine precursor aminoimidazole-carboxamide (see Figure 35–3). The drug inhibits the synthesis of guanine nucleotides, which are essential for both DNA and RNA viruses. It also inhibits the 5´ capping of viral mRNA. Ribavirin aerosol is used clinically to treat pneumonitis caused by respiratory syncytial virus (RSV) in infants and to treat severe influenza B infections. Ribavirin is also used in combination with α-interferon (peginterferon) for the treatment of hepatitis C.
INHIBITION OF INTEGRASE
Raltegravir (Isentress) is an integrase inhibitor (i.e., it blocks the HIV-encoded integrase that mediates the integration of the newly synthesized viral DNA into host cell DNA). Two additional integrase inhibitors are available: dolutegravir (Tivicay) and elvitegravir (Stribild).
INHIBITION OF CLEAVAGE OF PRECURSOR POLYPEPTIDES (PROTEASE INHIBITORS)
Inhibitors of Human Immunodeficiency Virus
Members of the protease inhibitor (PI) class of drugs, such as saquinavir (Invirase, Fortovase), indinavir (Crixivan), ritonavir (Norvir), lopinavir/ritonavir (Kaletra), atazanavir (Reyataz), tipranavir (Aptivus), amprenavir (Agenerase) and its prodrug fosamprenavir (Lexiva), darunavir (Prezista), and nelfinavir (Viracept), inhibit the protease encoded by HIV (Figure 35–5). The protease cleaves the gag and pol precursor polypeptides to produce several nucleocapsid proteins (e.g., p24) and enzymatic proteins (e.g., reverse transcriptase) required for viral replication. These inhibitors contain peptide bonds that bind to the active site of the viral protease, thereby preventing the protease from cleaving the viral precursor. These drugs inhibit production of infectious virions but do not affect the proviral DNA and therefore do not cure the infection.
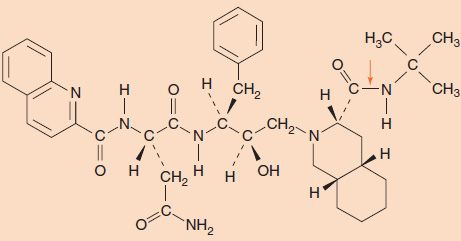
FIGURE 35–5 Structure of the protease inhibitor saquinavir. Note the presence of several peptide bonds, which interact with the active site of the protease. An arrow indicates one of the peptide bonds.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


