Antiviral Agents (Nonretroviral)
Viruses are simple microorganisms that consist of either double- or single-stranded DNA or RNA enclosed in a protein coat called a capsid. Some viruses also possess a lipid envelope derived from the infected host cell, which, like the capsid, may contain antigenic glycoproteins. Effective antiviral agents inhibit virus-specific replicative events or preferentially inhibit virus-directed rather than host cell–directed nucleic acid or protein synthesis (Table 58–1). Host cell molecules that are essential to viral replication also offer targets for intervention. Figure 58–1 gives a schematic diagram of the replicative cycle of typical DNA and RNA viruses.
Table 58–1
Stages of Virus Replication and Possible Targets of Action of Antiviral Agents

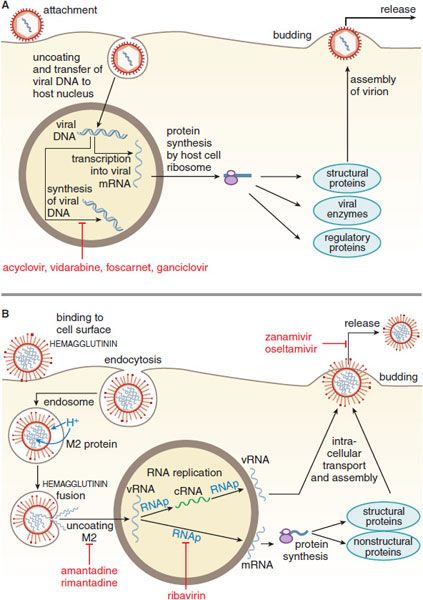
Figure 58–1 Replicative cycles of DNA (A) and RNA (B) viruses. The replicative cycles of herpesvirus (A) and influenza (B) are examples of DNA-encoded and RNA-encoded viruses, respectively. Sites of action of antiviral agents also are shown. cDNA, complementary DNA; cRNA, complementary RNA; DNAp, DNA polymerase; mRNA, messenger RNA; RNAp, RNA polymerase; vRNA, viral RNA. The symbol  indicates a block to virus growth. A. Replicative cycles of herpes simplex virus, a DNA virus, and the probable sites of action of antiviral agents. Herpesvirus replication is a regulated multistep process. After infection, a small number of immediate-early genes are transcribed; these genes encode proteins that regulate their own synthesis and are responsible for synthesis of early genes involved in genome replication, such as thymidine kinases, DNA polymerases, etc. After DNA replication, the bulk of the herpesvirus genes (called late genes) are expressed and encode proteins that either are incorporated into or aid in the assembly of progeny virions. B. Replicative cycles of influenza, an RNA virus, and the loci for effects of antiviral agents. The mammalian cell shown is an airway epithelial cell. The M2 protein of influenza virus allows an influx of hydrogen ions into the virion interior, which in turn promotes dissociation of the RNP (ribonuclear protein) segments and release into the cytoplasm (uncoating). Influenza virus mRNA synthesis requires a primer cleared from cellular mRNA and used by the viral RNAp complex. The neuraminidase inhibitors zanamivir and oseltamivir specifically inhibit release of progeny virus.
indicates a block to virus growth. A. Replicative cycles of herpes simplex virus, a DNA virus, and the probable sites of action of antiviral agents. Herpesvirus replication is a regulated multistep process. After infection, a small number of immediate-early genes are transcribed; these genes encode proteins that regulate their own synthesis and are responsible for synthesis of early genes involved in genome replication, such as thymidine kinases, DNA polymerases, etc. After DNA replication, the bulk of the herpesvirus genes (called late genes) are expressed and encode proteins that either are incorporated into or aid in the assembly of progeny virions. B. Replicative cycles of influenza, an RNA virus, and the loci for effects of antiviral agents. The mammalian cell shown is an airway epithelial cell. The M2 protein of influenza virus allows an influx of hydrogen ions into the virion interior, which in turn promotes dissociation of the RNP (ribonuclear protein) segments and release into the cytoplasm (uncoating). Influenza virus mRNA synthesis requires a primer cleared from cellular mRNA and used by the viral RNAp complex. The neuraminidase inhibitors zanamivir and oseltamivir specifically inhibit release of progeny virus.
DNA viruses include poxviruses (smallpox), herpesviruses (chickenpox, shingles, oral and genital herpes), adenoviruses (conjunctivitis, sore throat), hepadnaviruses (hepatitis B virus [HBV]), and papillomaviruses (warts). Most DNA viruses enter the host cell nucleus, where the viral DNA is transcribed into mRNA by host cell polymerase; mRNA is translated in the usual host cell fashion into virus-specific proteins. Poxviruses are an exception; they carry their own RNA polymerase and replicate in the host cell cytoplasm.
For RNA viruses, the replication strategy relies either on enzymes in the virion to synthesize mRNA or has the viral RNA serving as its own mRNA. The mRNA is translated into various viral proteins, including RNA polymerase, which directs the synthesis of more viral mRNA and genomic RNA. Most RNA viruses complete their replication in the host cell cytoplasm, but some, such as influenza, are transcribed in the host cell nucleus. Examples of RNA viruses include rubella virus (German measles), rhabdoviruses (rabies), picornaviruses (poliomyelitis, meningitis, colds, hepatitis A), arenaviruses (meningitis, Lassa fever), flaviviruses (West Nile meningoencephalitis, yellow fever, hepatitis C), orthomyxoviruses (influenza), paramyxoviruses (measles, mumps), and coronaviruses (colds, severe acute respiratory syndrome [SARS]). Retroviruses are RNA viruses that include human immunodeficiency virus (HIV); chemotherapy for retro-viruses is described in Chapter 59.
Table 58–2 summarizes currently approved drugs for nonretroviral infections. Their pharmacological properties are presented below, class by class, as listed in the table.
Table 58–2
Nomenclature of Antiviral Agents
ANTI-HERPESVIRUS AGENTS
Herpes simplex virus type 1 (HSV-1) typically causes diseases of the mouth, face, skin, esophagus, or brain. Herpes simplex virus type 2 (HSV-2) usually causes infections of the genitals, rectum, skin, hands, or meninges. Both cause serious infections in neonates. Acyclovir is the prototype of a group of antiviral agents that are nucleoside congeners that are phosphorylated intracellularly by a viral kinase and subsequently by host cell enzymes to become inhibitors of viral DNA synthesis. Related agents include penciclovir and ganciclovir.
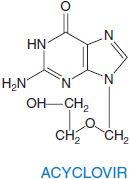
ACYCLOVIR AND VALACYCLOVIR. Acyclovir is an acyclic guanine nucleoside analog that lacks the 2′ and 3′positions normally supplied by ribose. Valacyclovir is the L-valyl ester prodrug of acyclovir.
Acyclovir’s clinical use is limited to herpesviruses. Acyclovir is most active against HSV-1 (0.02-0.9 μg/mL), approximately half as active against HSV-2 (0.03-2.2 μg/mL), a tenth as potent against varicella zoster virus (VZV; 0.8-4.0 μg/mL) and Epstein-Barr virus (EBV), and least active against cytomegalovirus (CMV) (generally >20 μg/mL) and human herpesvirus 6 (HHV-6). Uninfected mammalian cell growth generally is unaffected by high acyclovir concentrations (>50 μg/mL).
THERAPEUTIC USES. In immunocompetent persons, the clinical benefits of acyclovir and valacyclovir are greater in initial HSV infections than in recurrent ones. These drugs are particularly useful in immunocompromised patients because these individuals experience both more frequent and more severe HSV and VZV infections. Because VZV is less susceptible than HSV to acyclovir, higher doses must be used for treating VZV infections. Oral valacyclovir is as effective as oral acyclovir in HSV infections and more effective for treating herpes zoster. Acyclovir is ineffective therapeutically in established cytomegalovirus (CMV) infections but ganciclovir is effective for CMV prophylaxis in immunocompromised patients. EBV-related oral hairy leukoplakia may improve with acyclovir. Oral acyclovir in conjunction with systemic corticosteroids appears beneficial in treating Bell palsy; valacyclovir is ineffective in acute vestibular neuritis. See the 12th edition of the parent text for details of dosage regimens for specific indications in treating HSV, VZV, and CMV.
MECHANISMS OF ACTION AND RESISTANCE. Acyclovir inhibits viral DNA synthesis via a mechanism outlined in Figure 58–2. Its selectivity of action depends on interaction with HSV thymidine kinase and DNA polymerase. Cellular uptake and initial phosphorylation are facilitated by HSV thymidine kinase. The affinity of acyclovir for HSV thymidine kinase is ~200 times greater than for the mammalian enzyme. Cellular enzymes convert the monophosphate to acyclovir triphosphate, which competes for endogenous dGTP. The immunosuppressive agent mycophenolate mofetil (see Chapter 35) potentiates the anti-herpes activity of acyclovir and related agents by depleting intracellular dGTP pools. Acyclovir triphosphate competitively inhibits viral DNA polymerases and, to a much lesser extent, cellular DNA polymerases. Acyclovir triphosphate also is incorporated into viral DNA, where it acts as a chain terminator because of the lack of a 3′-hydroxyl group. By a mechanism termed suicide inactivation, the terminated DNA template containing acyclovir binds the viral DNA polymerase and leads to its irreversible inactivation.

Figure 58–2 Mechanism of action of acyclovir in cells infected by herpes simplex virus. A herpes simplex virion is shown attaching to a susceptible host cell, fusing its envelope with the cell membrane, and releasing naked capsids that deliver viral DNA into the nucleus, where it initiates synthesis of viral DNA. Acyclovir molecules entering the cell are converted to acyclovir monophosphate by virus-induced thymidine kinase. Host-cell enzymes add 2 more phosphates to form acyclovir triphosphate, which is transported into the nucleus. After the herpes DNA polymerase cleaves pyrophosphate from acyclovir triphosphate (indicated by the red arrow in the inset), viral DNA polymerase inserts acyclovir monophosphate rather than 2′-deoxyguanosine monophosphate into the viral DNA (indicated by black arrows in the inset). Further elongation of the chain is impossible because acyclovir monophosphate lacks the 3′ hydroxyl group necessary for the insertion of an additional nucleotide, and the exonuclease associated with the viral DNA polymerase cannot remove the acyclovir moiety. In contrast, ganciclovir and penciclovir have a 3′ hydroxyl group; therefore, further synthesis of viral DNA is possible in the presence of these drugs. Foscarnet acts at the pyrophosphate-binding site of viral DNA polymerase and prevents cleavage of the pyrophosphate from nucleoside triphosphates, thus stalling further primer template extension. Red lines [III] indicate hydrogen bonding of the base pairs. (Adapted from Balfour HH. Antiviral drugs. N Engl J Med, 1999;340:1255–1268.)
Acyclovir resistance in HSV can result from impaired production of viral thymidine kinase, altered thymidine kinase substrate specificity (e.g., phosphorylation of thymidine but not acyclovir), or altered viral DNA polymerase. Alterations in viral enzymes are caused by point mutations and base insertions or deletions in the corresponding genes. Resistant variants are present in native virus populations and in isolates from treated patients. The most common resistance mechanism in clinical HSV isolates is absent or deficient viral thymidine kinase activity; viral DNA polymerase mutants are rare. Phenotypic resistance typically is defined by in vitro inhibitory concentrations of >2-3 μg/mL, which predict failure of therapy in immunocompromised patients. Acyclovir resistance in VZV isolates is caused by mutations in VZV thymidine kinase and less often by mutations in viral DNA polymerase.
ADME. The oral bioavailability of acyclovir is ~10-30% and decreases with increasing dose. Valacyclovir is converted rapidly and virtually completely to acyclovir after oral administration. This conversion is thought to result from first-pass intestinal and hepatic metabolism through enzymatic hydrolysis. Unlike acyclovir, valacyclovir is a substrate for intestinal and renal peptide transporters. The oral bioavailability of acyclovir increases ~70% following valacyclovir administration. Peak plasma concentrations of valacyclovir are only 4% of acyclovir levels. Less than 1% of an administered dose of valacyclovir is recovered in the urine; most is eliminated as acyclovir. Acyclovir distributes widely in body fluids, including vesicular fluid, aqueous humor, and cerebrospinal fluid (CSF). Compared with plasma, salivary concentrations are low, and vaginal secretion concentrations vary widely. Acyclovir is concentrated in breast milk, amniotic fluid, and placenta. Newborn plasma levels are similar to maternal ones. Percutaneous absorption of acyclovir after topical administration is low. The elimination t1/2 of acyclovir is ~2.5 h (range: 1.5-6 h) in adults with normal renal function. The elimination t1/2 of acyclovir is ~4 h in neonates and increases to 20 h in anuric patients. Renal excretion is the principal route of elimination.
UNTOWARD EFFECTS. Acyclovir generally is well tolerated. Topical acyclovir in a polyethylene glycol base may cause mucosal irritation and transient burning when applied to genital lesions. Oral acyclovir has been associated infrequently with nausea, diarrhea, rash, or headache and very rarely with renal insufficiency or neurotoxicity. Valacyclovir also may be associated with headache, nausea, diarrhea, nephrotoxicity, and CNS symptoms (confusion, hallucinations). Uncommon side effects include severe thrombocytopenic syndromes, sometimes fatal, in immunocompromised patients. Acyclovir has been associated with neutropenia in neonates. The principal dose-limiting toxicities of intravenous acyclovir are renal insufficiency and CNS side effects. Nephrotoxicity usually resolves with drug cessation and volume expansion. Hemodialysis may be useful in severe cases. Severe somnolence and lethargy may occur with combinations of zidovudine (see Chapter 59) and acyclovir. Concomitant cyclosporine and probably other nephrotoxic agents enhance the risk of nephrotoxicity. Probenecid decreases the acyclovir renal clearance and prolongs the elimination t1/2. Acyclovir may decrease the renal clearance of other drugs eliminated by active renal secretion, such as methotrexate.
CIDOFOVIR. Cidofovir is a cytidine nucleotide analog with inhibitory activity against human herpes, papilloma, polyoma, pox, and adenoviruses.
Because cidofovir is a phosphonate that is phosphorylated by cellular but not viral enzymes, it inhibits acyclovir-resistant thymidine kinase (TK)–deficient or TK-altered HSV or VZV strains, ganciclovir-resistant CMV strains with UL97 mutations (but not those with DNA polymerase mutations), and some foscarnet-resistant CMV strains. Cidofovir synergistically inhibits CMV replication in combination with ganciclovir or foscarnet.
THERAPEUTIC USES. Intravenous cidofovir is approved for the treatment of CMV retinitis in HIV-infected patients. Intravenous cidofovir has been used for treating acyclovir-resistant mucocutaneous HSV infection, adenovirus disease in transplant recipients, and extensive molluscum contagiosum in HIV patients. Reduced doses without probenecid may be beneficial in BK virus nephropathy in renal transplant patients. Topical cidofovir gel eliminates virus shedding and lesions in some HIV-infected patients with acyclovir-resistant mucocutaneous HSV infections and has been used in treating anogenital warts and molluscum contagiosum in immunocompromised patients and cervical intraepithelial neoplasia in women. Intralesional cidofovir induces remissions in adults and children with respiratory papillomatosis. See the 12th edition of the parent text for details of dosage regimens for specific indications.
MECHANISMS OF ACTION AND RESISTANCE. Cidofovir inhibits viral DNA synthesis by slowing and eventually terminating chain elongation. Cidofovir is metabolized to its active diphosphate form by cellular enzymes; the levels of phosphorylated metabolites are similar in infected and uninfected cells. The diphosphate acts as both a competitive inhibitor with respect to dCTP and as an alternative substrate for viral DNA polymerase.
Cidofovir resistance in CMV is due to mutations in viral DNA polymerase. Low-level resistance to cidofovir develops in up to ~30% of retinitis patients by 3 months of therapy. Highly ganciclovir-resistant CMV isolates that possess DNA polymerase and UL97 kinase mutations are resistant to cidofovir, and prior ganciclovir therapy may select for cidofovir resistance. Some foscarnet-resistant CMV isolates show cross-resistance to cidofovir, and triple-drug-resistant variants with DNA polymerase mutations occur.
ADME. Cidofovir has very low oral bioavailability. Penetration into the CSF is low. Topical cidofovir gel may result in low plasma concentrations (<0.5 μg/mL) in patients with large mucocutaneous lesions. Plasma levels after intravenous dosing decline in a biphasic pattern with a terminal t1/2 that averages 2.6 h. The active form, cidofovir diphosphate, has a prolonged intracellular t1/2 and competitively inhibits CMV and HSV DNA polymerases at concentrations one-eighth to one six-hundredth of those required to inhibit human DNA polymerases. A phosphocholine metabolite also has a long intracellular t1/2 (~87 h) and may serve as an intracellular reservoir of drug. The prolonged intracellular t1/2 of cidofovir diphosphate allows infrequent dosing regimens. Cidofovir is cleared by the kidney via glomerular filtration and tubular secretion. Over 90% of the dose is recovered unchanged in the urine. Probenecid blocks tubular transport of cidofovir and reduces renal clearance and associated nephrotoxicity. Elimination relates linearly to creatinine clearance; the t1/2 increases to 32.5 h in patients on chronic ambulatory peritoneal dialysis (CAPD). Hemodialysis removes >50% of the administered dose.
UNTOWARD EFFECTS. Nephrotoxicity is the principal dose-limiting side effect of intravenous cidofovir. Concomitant oral probenecid and saline prehydration reduce the risk of renal toxicity; however, probenecid alters renal clearance of many agents, albeit not of cidofidir. For example, probenecid alters zidovudine pharmacokinetics such that zidovudine doses should be reduced when probenecid is present, as should the doses of other drugs whose renal secretion probenecid inhibits (e.g., β-lactam antibiotics, nonsteroidal anti-inflammatory drugs [NSAIDs], acyclovir, lorazepam, furosemide, methotrexate, theophylline, and rifampin). On maintenance doses of 5 mg/kg every 2 weeks, up to 50% of patients develop proteinuria, 10-15% show an elevated serum creatinine concentration, and 15-20% develop neutropenia. Anterior uveitis that is responsive to topical corticosteroids and cycloplegia occurs commonly and low intraocular pressure occurs infrequently with intravenous cidofovir. Administration with food and pretreatment with antiemetics, antihistamines, and/or acetaminophen may improve tolerance. Concurrent nephrotoxic agents are contraindicated, and at least 7 days should elapse before initiation of cidofovir treatment is recommended after prior exposure to aminoglycosides, intravenous pentamidine, amphotericin B, foscarnet, NSAID, or contrast dye. Cidofovir and oral ganciclovir are poorly tolerated in combination at full doses.
Topical application of cidofovir is associated with dose-related application-site reactions (e.g., burning, pain, and pruritus) in up to one-third of patients and occasionally ulceration. Cidofovir is considered a potential human carcinogen. It may cause infertility and is classified as pregnancy Category C.
FAMCICLOVIR AND PENCICLOVIR. Famciclovir is the diacetyl ester prodrug of 6-deoxy penciclovir and lacks intrinsic antiviral activity. Penciclovir is an acyclic guanine nucleoside analog. Penciclovir is similar to acyclovir in its spectrum of activity and potency against HSV and VZV. It also is inhibitory for HBV.
THERAPEUTIC USES. Oral famciclovir, topical penciclovir, and intravenous penciclovir are approved for managing HSV and VZV infections. See the 12th edition of the parent text for details of dosage regimens for specific indications.
MECHANISMS OF ACTION AND RESISTANCE. Penciclovir is an inhibitor of viral DNA synthesis. In HSV- or VZV-infected cells, penciclovir is phosphorylated initially by viral TK. Penciclovir triphosphate is a competitive inhibitor of viral DNA polymerase (see Figure 58–2). Although penciclovir triphosphate is approximately one one-hundredth as potent as acyclovir triphosphate in inhibiting viral DNA polymerase, it is present in infected cells at much higher concentrations and for more prolonged periods. The prolonged intracellular t1/2 of penciclovir triphosphate, 7-20 h, is associated with prolonged antiviral effects. Because penciclovir has a 3′-hydroxyl group, it is not an obligate chain terminator but does inhibit DNA elongation. Resistance during clinical use is low. TK–deficient, acyclovir-resistant herpes viruses are cross-resistant to penciclovir.
ADME. Oral penciclovir has low (<5%) bioavailability. In contrast, famciclovir is well absorbed orally (bioavailability ~75%) and is converted rapidly to penciclovir by deacetylation of the side chain and oxidation of the purine ring during and following absorption. Food slows absorption but does not reduce overall bioavailability. The plasma elimination t1/2 of penciclovir averages ~2 h, and >90% is excreted unchanged in the urine. Following oral famciclovir administration, nonrenal clearance accounts for ~10% of each dose, primarily through fecal excretion, but penciclovir (60% of dose) and its 6-deoxy precursor (<10% of dose) are eliminated primarily in the urine. The plasma t1/2 averages 9.9 h in renal insufficiency (Clcr 30 mL/min); hemodialysis efficiently removes penciclovir.
UNTOWARD EFFECTS. Oral famciclovir is associated with headache, diarrhea, and nausea. Urticaria, rash, and hallucinations or confusional states (predominantly in the elderly) have been reported. Topical penciclovir (~1%) rarely is associated with local reactions. The short-term tolerance of famciclovir is comparable with that of acyclovir. Penciclovir is mutagenic at high concentrations. Long-term administration (1 year) does not affect spermatogenesis in men. Safety during pregnancy has not been established.
FOMIVIRSEN. Fomivirsen, a 21-base phosphorothioate oligonucleotide, provides antisense therapy.
The drug is complementary to the messenger RNA sequence for the major immediate-early transcriptional region of CMV and inhibits CMV replication through sequence-specific and nonspecific mechanisms, including inhibition of virus binding to cells. Fomivirsen is active against CMV strains resistant to ganciclovir, foscarnet, and cidofovir. Fomivirsen is given by intravitreal injection in the treatment of CMV retinitis for patients intolerant of or unresponsive to other therapies. Following injection, it is cleared slowly from the vitreous (t1/2 55 h) through distribution to the retina and probable exonuclease digestion. In HIV-infected patients with refractory, sight-threatening CMV retinitis, fomivirsen injections (330 μg weekly for 3 weeks and then every 2 weeks or on days 1 and 15 followed by monthly) significantly delay time to retinitis progression. Ocular side effects include iritis in up to one-quarter of patients, which can be managed with topical corticosteroids; vitritis; cataracts; and increases in intraocular pressure in 15-20% of patients. Recent cidofovir use may increase the risk of inflammatory reactions. The drug is no longer available in the U.S.
FOSCARNET. Foscarnet (trisodium phosphonoformate) is an inorganic pyrophosphate analog that is inhibitory for all herpesviruses and HIV.
THERAPEUTIC USES. Intravenous foscarnet is effective for treatment of CMV retinitis, including ganciclovir-resistant infections, other types of CMV infection, and acyclovir-resistant HSV and VZV infections. Foscarnet is poorly soluble in aqueous solutions and requires large volumes for administration. See the 12th edition of the parent text for details of dosage regimens for specific indications.
MECHANISMS OF ACTION AND RESISTANCE. Foscarnet inhibits viral nucleic acid synthesis by interacting directly with herpesvirus DNA polymerase or HIV reverse transcriptase (see Figures 58–1A and 58–2). Foscarnet has ~100-fold greater inhibitory effects against herpesvirus DNA polymerases than against cellular DNA polymerase-α. Herpesviruses resistant to foscarnet have point mutations in the viral DNA polymerase.
ADME. Oral bioavailability of foscarnet is low. Vitreous levels approximate those in plasma; CSF levels average 66% of those in plasma at steady state. Over 80% of foscarnet is excreted unchanged in the urine. Dose adjustments are indicated for small decreases in renal function. Plasma elimination has initial bimodal half-lives totaling 4-8 h and a prolonged terminal elimination t1/2 averaging 3-4 days. Sequestration in bone with gradual release accounts for the fate of an estimated 10-20% of a given dose. Foscarnet is cleared efficiently by hemodialysis (~50% of a dose).
UNTOWARD EFFECTS. Major dose-limiting toxicities are nephrotoxicity and symptomatic hypocalcemia. Increases in serum creatinine occur in up to one-half of patients but are generally reversible after cessation. High doses, rapid infusion, dehydration, prior renal insufficiency, and concurrent nephrotoxic drugs are risk factors. Saline loading may reduce the risk of nephrotoxicity. Foscarnet is highly ionized at physiological pH, and metabolic abnormalities are very common. These include increases or decreases in Ca2+ and phosphate, hypomagnesemia, and hypokalemia. Concomitant intravenous pentamidine administration increases the risk of symptomatic hypocalcemia. CNS side effects include headache (25%), tremor, irritability, seizures, and hallucinosis. Other reported side effects are generalized rash, fever, nausea or emesis, anemia, leukopenia, abnormal liver function tests, electrocardiographic changes, infusion-related thrombophlebitis, and painful genital ulcerations. Topical foscarnet may cause local irritation and ulceration, and oral foscarnet may cause GI disturbance. Preclinical studies indicate that high foscarnet concentrations are mutagenic. Safety in pregnancy or childhood is uncertain.
GANCICLOVIR AND VALGANCICLOVIR. Ganciclovir is an acyclic guanine nucleoside analog that is similar in structure to acyclovir. Valganciclovir is the L-valyl ester prodrug of ganciclovir. Ganciclovir has inhibitory activity against all herpesviruses and is especially active against CMV.
THERAPEUTIC USES.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


