Antiretroviral Agents and Treatment of HIV Infection
The pharmacotherapy of HIV infection is a rapidly moving field. Three-drug combinations are the minimum standard of care for this infection, so current agents constitute several thousand possible regimens. Knowing the essential features of the pathophysiology of this disease and how chemotherapeutic agents affect the virus and the host is critical in developing a rational approach to therapy. Unique features of this drug class include the need for lifelong administration to control virus replication and the possibility of rapid emergence of permanent drug resistance if these agents are not used properly.
PATHOGENESIS OF HIV-RELATED DISEASE
Human immunodeficiency viruses (HIVs) are lentiviruses, a family of retroviruses evolved to establish chronic persistent infection with gradual onset of clinical symptoms. Replication is constant following infection, and although some infected cells may harbor nonreplicating virus for years, in the absence of treatment there generally is no true period of viral latency following infection. Humans and nonhuman primates are the only natural hosts for these viruses.
There are 2 major families of HIV. Most of the epidemic involves HIV-1; HIV-2 is more closely related to simian immunodeficiency virus (SIV) and is concentrated in western Africa. HIV-1 is genetically diverse, with at least 5 distinct subfamilies or clades. HIV-1 and HIV-2 have similar sensitivity to most antiretroviral drugs, although the non-nucleoside reverse transcriptase inhibitors (NNRTIs) are HIV-1-specific and have no activity against HIV-2.
VIRUS STRUCTURE. HIV is a typical retrovirus with a small RNA genome of 9300 base pairs. Two copies of the genome are contained in a nucleocapsid core surrounded by a lipid bilayer, or envelope, that is derived from the host cell plasma membrane (Figure 59–1). The viral genome encodes 3 major open reading frames: gag encodes a polyprotein that is processed to release the major structural proteins of the virus; pol overlaps gag and encodes 3 important enzyme activities—an RNA-dependent DNA polymerase or reverse transcriptase with RNAase activity, protease, and the viral integrase; and env encodes the large transmembrane envelope protein responsible for cell binding and entry. Several small genes encode regulatory proteins that enhance virion production or combat host defenses. These include tat, rev, nef, and vpr.
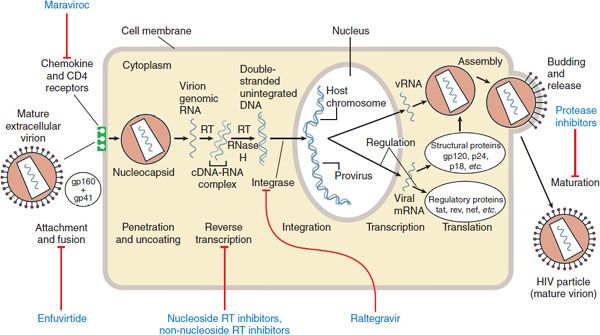
Figure 59–1 Replicative cycle of HIV-1 and sites of action of available antiretroviral agents. Available antiretroviral agents are shown in blue. cDNA, complementary DNA; gp120 + gp41, extracellular and intracellular domains, respectively, of envelope glycoprotein; mRNA, messenger RNA; RNase H, ribonuclease H; RT, reverse transcriptase. (Adapted from Hirsch MS, D′Aquila RT. Therapy for human immunodeficiency virus infection. N Engl J Med, 1993; 328:1686–1695.)
VIRUS LIFE CYCLE (see Figure 59–1). HIV tropism is controlled by the envelope protein gp160 (env). The major target for env binding is the CD4 receptor present on lymphocytes and macrophages, although cell entry also requires binding to a coreceptor, generally the chemokine receptor CCR5 or CXCR4. CCR5 is present on macrophage lineage cells. Most infected individuals harbor predominantly the CCR5-tropic virus; HIV with this tropism is responsible for nearly all naturally acquired infections. A shift from CCR5 to CXCR4 utilization is associated with advancing disease, and the increased affinity of HIV-1 for CXCR4 allows infection of T-lymphocyte lines. A phenotypic switch from CCR5 to CXCR4 heralds accelerated loss of CD4+ helper T cells and increased risk of immunosuppression. Whether coreceptor switch is a cause or a consequence of advancing disease is still unknown, but it is possible to develop clinical AIDS without this switch.
The gp41 domain of env controls the fusion of the virus lipid bilayer with that of the host cell. Following fusion, full-length viral RNA enters the cytoplasm, where it undergoes replication to a short-lived RNADNA duplex; the original RNA is degraded by RNase H to allow creation of a full-length double-stranded DNA copy of the virus. Because the HIV reverse transcriptase is error prone and lacks a proofreading function, mutation is quite frequent and occurs at ~3 bases out of every full-length (9300-base-pair) replication. Virus DNA is transported into the nucleus, where it is integrated into a host chromosome by the viral integrase in a random or quasi-random location.
Following integration, the virus may remain quiescent, not producing RNA or protein but replicating as the cell divides. When a cell that harbors the virus is activated, viral RNA and proteins are produced. Structural proteins assemble around full-length genomic RNA to form a nucleocapsid. The envelope and structural proteins assemble at the cell surface, concentrating in cholesterol-rich lipid rafts. The nucleocapsid cores are directed to these sites and bud through the cell membrane, creating new enveloped HIV particles containing 2 complete single-stranded RNA genomes. Reverse transcriptase is incorporated into virus particles so replication can begin immediately after the virus enters a new cell.
HOW THE VIRUS CAUSES DISEASE. Sexual acquisition of HIV infection is likely mediated by one or, at most, a handful of infectious virus particles. Soon after infection, there is a rapid burst of replication peaking at 2-4 weeks, with ≥109 cells becoming infected. This peak is associated with a transient dip in the number of peripheral CD4+ (helper) T lymphocytes. As a result of new host immune responses and target cell depletion, the number of infectious virions as reflected by the plasma HIV RNA concentration (also known as viral load) declines to a quasi-steady state. This set point reflects the interplay between host immunity and the pathogenicity of the infecting virus. In the average infected individual, several billion infectious virus particles are produced every few days.
Eventually, the host CD4+ T lymphocyte count begins a steady decline, accompanied by a rise in the plasma HIV RNA concentration. Once the peripheral CD4 cell count falls below 200 cells/mm3, there is an increasing risk of opportunistic diseases and ultimately death. Sexual acquisition of CCR5-tropic HIV-1 is associated with a median time to clinical AIDS of 8-10 years. Some patients, termed long-term nonprogressors, can harbor HIV for more than 2 decades without significant decline in peripheral CD4 cell count or clinical immunosuppression; this may reflect a combination of favorable host immunogenetics and immune responses.
An important question relevant to treatment is whether HIV disease is a consequence of CD4+ lymphocyte depletion alone. Most natural history data suggest that this is true. Regardless, successful therapy is based on inhibition of HIV replication; interventions designed specifically to boost the host immune response without exerting a direct antiviral effect have had no reliable clinical benefit.
PRINCIPLES OF HIV CHEMOTHERAPY
Current treatment assumes that all aspects of disease derive from the direct toxic effects of HIV on host cells, mainly CD4+ T lymphocytes. The goal of therapy is to suppress virus replication as much as possible for as long as possible. The current standard of care is to use at least 3 drugs simultaneously for the entire duration of treatment.
Current guidelines in the U.S. recommend starting therapy in all those with a CD4 count of ≤350 cells/mm3. Treatment is also recommended for HIV-infected pregnant women, those with HIV nephropathy, and those with concurrent hepatitis B virus infection requiring treatment regardless of CD4 count. Increasing evidence supports the value of antiretroviral therapy in preventing transmission of the virus from person to person. In the foreseeable future, treatment may be recommended for all infected adults and children.
Drug resistance is a key problem. There is a high likelihood that all untreated infected individuals harbor viruses with single-amino-acid mutations conferring some degree of resistance to every known antiretroviral drug because of the high mutation rate of HIV and the tremendous number of infectious virions. Thus, a combination of active agents is required to prevent drug resistance, analogous to strategies employed in the treatment of tuberculosis (see Chapter 56). Intentional drug holidays, also known as structured treatment interruptions, allow the virus to replicate anew, increase the risk of drug resistance and disease progression, and are not recommended.
The expected outcome of initial therapy in a previously untreated patient is an undetectable viral load (plasma HIV RNA <50 copies/mL) within 24 weeks of starting treatment. Mathematical models of HIV replication suggested that 3 is the minimum number of agents required to guarantee effective long-term suppression of HIV replication without resistance. In treatment-naive patients, a regimen containing a non-nucleoside plus 2 nucleoside reverse transcriptase inhibitors is as effective as a regimen containing an additional nucleoside, indicating the equivalence of these 3-drug and 4-drug regimens. Four or more drugs may be used simultaneously in pretreated patients harboring drug-resistant virus, but the number of agents a patient can take is limited by toxicity and inconvenience.
Failure of an antiretroviral regimen is defined as a persistent increase in plasma HIV RNA concentrations in a patient with previously undetectable virus, despite continued treatment with that regimen. This indicates resistance to 1 or more drugs in the regimen and necessitates a change in treatment. The selection of new agents is therefore informed by the patient’s treatment history and viral resistance testing. Treatment failure generally requires implementation of a completely new combination of drugs. Adding a single active agent to a failing regimen is functional monotherapy if the patient is resistant to all drugs in the regimen. The risk of failing a regimen depends on the percentage of prescribed doses taken during any given period of treatment.
As antiretroviral therapy becomes more effective and easier to take, long-term toxicity of these drugs is of greater concern. An important consequence of long-term therapy is the development of a metabolic syndrome (HIV lipodystrophy syndrome) characterized by insulin resistance, fat redistribution, and hyperlipidemia, that occurs in 10-40% of treated patients. Clinical features include peripheral fat wasting (lipoatrophy), central fat accumulation including enlarged breasts and buffalo hump, insulin resistance and hyperglycemia, and elevations in serum cholesterol and triglycerides. Lipodystrophy has been associated with an increased risk of myocardial infarction in virologically controlled patients. A potential concern that applies to all protease inhibitors and NNRTIs is clinically significant pharmacokinetic drug interactions. All agents in these 2 drug classes can act as inhibitors and/or inducers of hepatic CYPs and other drug metabolizing enzymes, as well as drug transport proteins.
An increasingly recognized complication of initiating antiretroviral therapy is accelerated inflammatory reaction to overt or subclinical opportunistic infections or malignancies. This immune reconstitution inflammatory syndrome (IRIS) is most commonly seen when initiating therapy in individuals with low CD4 counts and/or advanced HIV disease. Infections commonly associated with IRIS include tuberculosis and other mycobacterial diseases, cryptococcosis, hepatitis virus infections, and Pneumocystis pneumonia.
Drugs Used to Treat HIV Infection
NUCLEOSIDE AND NUCLEOTIDE REVERSE TRANSCRIPTASE INHIBITORS
The HIV-encoded, RNA-dependent DNA polymerase, also called reverse transcriptase, converts viral RNA into proviral DNA that is then incorporated into a host cell chromosome. Available inhibitors of this enzyme are either nucleoside/nucleotide analogs or non-nucleoside inhibitors (Table 59–1). Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) prevent infection of susceptible cells but do not eradicate the virus from cells that already harbor integrated proviral DNA. Nearly all patients starting antiretroviral treatment do so with at least 1 agent from this class. Figure 59–2 shows the mechanism of action of NRTIs, which involves phosphorylation by host cells to the active inhibitory form.
Table 59–1
Antiretroviral Agents Approved for Use in the U.S.
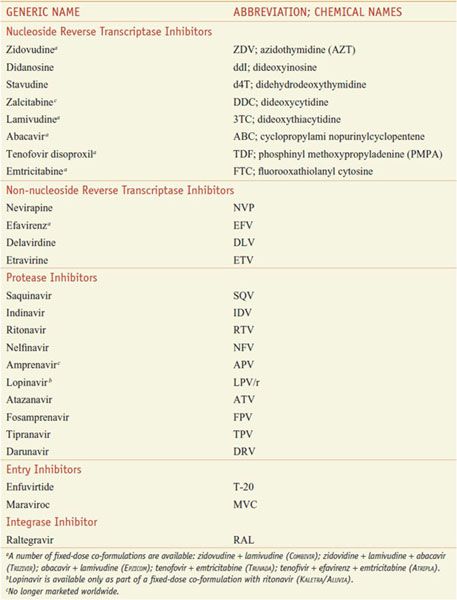
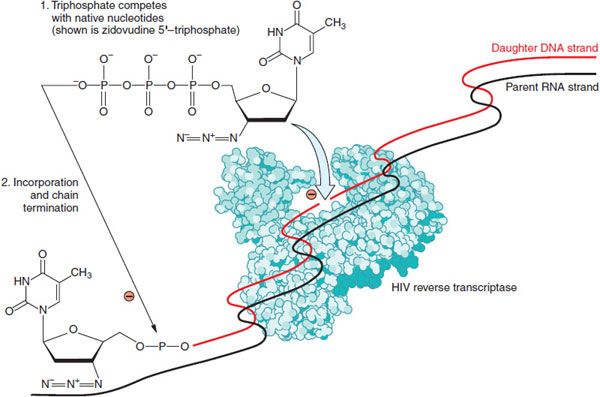
Figure 59–2 Mechanism of nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). Zidovudine is depicted; Table 59–1 lists other agents in the NRTI class. Nucleoside and nucleotide analogs must enter cells and be phosphorylated to generate synthetic substrates for reverse transcriptase. The fully phosphorylated analogs block replication of the viral genome both by competitively inhibiting incorporation of native nucleotides and by terminating elongation of nascent proviral DNA because they lack a 3′-hydroxyl group.
All but 1 of the drugs in this class are nucleosides that must be triphosphorylated at the 5′-hydroxyl to exert activity. The sole exception, tenofovir, is a nucleotide monophosphate analog that requires 2 additional phosphates to acquire full activity. These compounds inhibit both HIV-1 and HIV-2, and several have broad-spectrum activity against other human and animal retroviruses; emtricitabine, lamivudine, and tenofovir are active against hepatitis B virus (HBV), and tenofovir also has activity against herpesviruses (see Chapter 58).
The selective toxicity of these drugs depends on their ability to inhibit the HIV reverse transcriptase without inhibiting host cell DNA polymerases. Although the intracellular triphosphates for all these drugs have low affinity for human DNA polymerase-α and polymerase-β, some do inhibit human DNA polymerase-γ, which is the mitochondrial enzyme. As a result, the important toxicities common to this class of drugs result in part from the inhibition of mitochondrial DNA synthesis. These toxicities include anemia, granulocytopenia, myopathy, peripheral neuropathy, and pancreatitis. Lactic acidosis with or without hepatomegaly and hepatic steatosis is a rare but potentially fatal complication seen with stavudine, zidovudine, and didanosine. Phosphorylated emtricitabine, lamivudine, and tenofovir have low affinity for DNA polymerase-γ and are largely devoid of mitochondrial toxicity.
Table 59–2 summarizes the pharmacokinetic properties of NRTIs approved for treating HIV infection. A notable pharmacological feature of these agents is the elimination of the intracellular nucleoside di- or tri-phosphate, which is the active form. In general, the phosphorylated anabolites are eliminated from cells much more gradually than the parent drug is eliminated from the plasma. As a result, available NRTIs are dosed once or twice daily.
Table 59–2
Pharmacokinetic Properties of Nucleoside Reverse Transcriptase Inhibitorsa
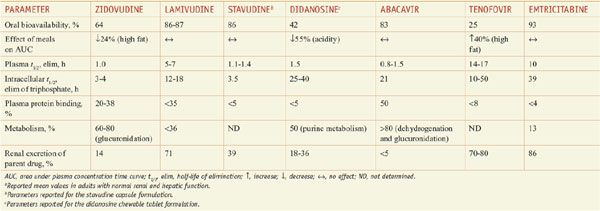
These drugs are not major substrates for hepatic CYPs. Pharmacokinetic drug interactions involving tenofovir and protease inhibitors are likely to be explained by inhibition of OATP drug transporters (see Chapter 5). High-level resistance to NRTIs, especially thymidine analogs, occurs slowly by comparison to non-NRTIs and first-generation protease inhibitors. High-level resistance can occur rapidly with lamivudine and emtricitabine. Cross-resistance is common but often confined to drugs having similar chemical structures. Several nucleoside analogs have favorable safety and tolerability profiles and are useful in suppressing the emergence of HIV isolates resistant to the more potent drugs in combination regimens.
ZIDOVUDINE. Zidovudine (AZT) is a synthetic thymidine analog (see structure in Figure 59–2) with potent activity against a broad spectrum of retroviruses including HIV-1, HIV-2, and human T-cell lymphotrophic viruses (HTLVs) I and II. Zidovudine is active in lymphoblastic and monocytic cell lines but has no impact on cells already infected with HIV. Zidovudine appears to be more active in activated than in resting lymphocytes because the phosphorylating enzyme, thymidine kinase, is S-phase-specific. Zidovudine is FDA-approved for the treatment of adults and children with HIV infection and for preventing mother-to-child transmission; it also is recommended for post-exposure prophylaxis in HIV-exposed healthcare workers. Zidovudine (RETROVIR) is marketed in oral tablets, capsules, and solution as well as a solution for intravenous injection. Zidovudine is available in coformulated tablets with lamivudine (COMBIVIR) or with lamivudine and abacavir (TRIZIVIR).
MECHANISMS OF ACTION AND RESISTANCE. Intracellular zidovudine is phosphorylated to zidovudine 5′-triphosphate. Zidovudine 5′-triphosphate terminates the elongation of proviral DNA because it is incorporated by reverse transcriptase into nascent DNA but lacks a 3′-hydroxyl group. The monophosphate competitively inhibits cellular thymidylate kinase, and this may reduce the amount of intracellular thymidine triphosphate. Zidovudine 5′-triphosphate only weakly inhibits cellular DNA polymerase-α but is a more potent inhibitor of mitochondrial polymerase-γ. Because the conversion of zidovudine 5′-monophosphate to diphosphate is very inefficient, high concentrations of the monophosphate accumulate inside cells and may serve as a precursor depot for formation of triphosphate. As a consequence, there is little correlation between extracellular concentrations of parent drug and intracellular concentrations of triphosphate, and higher plasma concentrations of zidovudine do not increase intracellular triphosphate concentrations proportionately.
Resistance to zidovudine is associated with mutations at reverse transcriptase codons 41, 44, 67, 70, 210, 215, and 219. These mutations are referred to as thymidine analog mutations (TAMs) because of their ability to confer cross-resistance to other thymidine analogs such as stavudine. The M184V substitution in the reverse transcriptase gene associated with the use of lamivudine or emtricitabine greatly restores sensitivity to zidovudine. The combination of zidovudine and lamivudine produces greater long-term suppression of plasma HIV RNA than does zidovudine alone.
ADME. Zidovudine is absorbed rapidly and reaches peak plasma concentrations within 1 h. Table 59–2 summarizes the drug’s pharmacokinetic profile, which is not altered significantly during pregnancy; drug concentrations in the newborn approach those of the mother. Parent drug crosses the blood-brain barrier relatively well and also is detectable in breast milk, semen, and fetal tissue.
UNTOWARD EFFECTS. Patients initiating zidovudine treatment often complain of fatigue, malaise, myalgia, nausea, anorexia, headache, and insomnia; these symptoms usually resolve within a few weeks. Erythrocytic macrocytosis is seen in ~90% of patients but usually is not associated with anemia. Chronic zidovudine administration has been associated with nail hyperpigmentation. Skeletal muscle myopathy can occur and is associated with depletion of mitochondrial DNA, most likely as a consequence of inhibition of DNA polymerase-γ. Serious hepatic toxicity, with or without steatosis and lactic acidosis, is rare but can be fatal.
PRECAUTIONS AND INTERACTIONS. Zidovudine is not a substrate or inhibitor of CYPs. However, probenecid, fluconazole, atovaquone, and valproic acid may increase plasma concentrations of zidovudine, probably through inhibition of glucuronosyl transferases. Zidovudine can cause bone marrow suppression and should be used cautiously in patients with preexisting anemia or granulocytopenia and in those taking other marrow-suppressive drugs. Stavudine and zidovudine compete for intracellular phosphorylation and should not be used concomitantly.
STAVUDINE. Stavudine (d4T) is a synthetic thymidine analog that is active in vitro against HIV-1 and HIV-2. Stavudine (ZERIT) is approved for use in HIV-infected adults and children, including neonates.
MECHANISMS OF ACTION AND RESISTANCE. Intracellular stavudine is sequentially phosphorylated stavudine 5′-triphosphate. Like zidovudine, stavudine is most potent in activated cells, probably because thymidine kinase, which produces the monophosphate, is an S-phase-specific enzyme. Stavudine resistance is seen most frequently with mutations at reverse transcriptase codons 41, 44, 67, 70, 210, 215, and 219, which are mutations associated with zidovudine resistance. Resistance mutations for stavudine appear to accumulate slowly. Cross-resistance to multiple nucleoside analogs has been reported following prolonged therapy.
ADME. Table 59–2 summarizes the agent’s PK data. Stavudine is well absorbed and reaches peak plasma concentrations within 1 h; bioavailability is not affected by food. The drug penetrates well into the CSF, achieving concentrations that are ~40% of those in plasma. Placental concentrations of stavudine are about half those of zidovudine. The drug undergoes active tubular secretion, and renal elimination accounts for ~40% of parent drug; thus, dose should be adjusted in patients with renal insufficiency.
UNTOWARD EFFECTS. The most common serious toxicity of stavudine is peripheral neuropathy. Although this may reflect mitochondrial toxicity, stavudine is a less potent inhibitor of DNA polymerase-γ than either didanosine or zalcitabine, suggesting that other mechanisms may be involved. Stavudine is also associated with a progressive motor neuropathy characterized by weakness and in some cases respiratory failure, similar to Guillain-Barré syndrome.
Lactic acidosis and hepatic steatosis are associated with stavudine use, and may be more common when stavudine and didanosine are combined. Acute pancreatitis is not highly associated with stavudine but is more common when stavudine is combined with didanosine than when didanosine is given alone. Of all nucleoside analogs, stavudine use is associated most strongly with fat wasting (lipoatrophy). Stavudine has fallen out of favor in the developed world largely because of this toxicity.
PRECAUTIONS AND INTERACTIONS. Stavudine is mainly renally cleared and is not subject to metabolic drug interactions. The incidence and severity of peripheral neuropathy may be increased when stavudine is combined with other neuropathic medications; thus, drugs such as ethambutol, isoniazid, phenytoin, and vincristine should be avoided. Combining stavudine with didanosine leads to increased risk and severity of peripheral neuropathy and potentially fatal pancreatitis; therefore, these 2 drugs should not be used together. Stavudine and zidovudine compete for intracellular phosphorylation and should not be used concomitantly.
LAMIVUDINE. Lamivudine is a cytidine analog reverse transcriptase inhibitor that is active against HIV-1, HIV-2, and HBV. Lamivudine (EPIVIR) is approved for HIV in adults and children ≥3 months of age. Lamivudine has been effective in combination with other antiretroviral drugs in both treatment-naive and experienced patients and is a common component of therapy, given its safety, convenience, and efficacy. Lamivudine (EPIVIR-HBV) is approved for treatment of chronic hepatitis B.
MECHANISMS OF ACTION AND RESISTANCE. Lamivudine enters cells by passive diffusion and is sequentially phosphorylated to lamivudine 5′-triphosphate, which is the active anabolite. Lamivudine has low affinity for human DNA polymerases, explaining its low toxicity to the host. High-level resistance to lamivudine occurs with single-amino-acid substitutions, M184V or M184I. These mutations can reduce in vitro sensitivity to lamivudine as much as 1000-fold. The M184V mutation restores zidovudine susceptibility in zidovudine-resistant HIV and also partially restores tenofovir susceptibility in tenofovir-resistant HIV harboring the K65R mutation. This effect may contribute to the sustained virologic benefits of zidovudine and lamivudine combination therapy.
ADME. Table 59–2 summarizes the PK parameters for this drug. Lamivudine is excreted primarily unchanged in the urine; dose adjustment is recommended for patients with a creatinine clearance <50 mL/min. Lamivudine freely crosses the placenta into the fetal circulation.
UNTOWARD EFFECTS. Lamivudine is one of the least toxic antiretroviral drugs. Neutropenia, headache, and nausea have been reported at higher than recommended doses. Pancreatitis has been reported in pediatric patients.
PRECAUTIONS AND INTERACTIONS. Because lamivudine also has activity against HBV, caution is warranted in using this drug in patients coinfected with HBV or in HBV-endemic areas: discontinuation of lamivudine may be associated with a rebound of HBV replication and exacerbation of hepatitis.
ABACAVIR. Abacavir (ZIAGEN), a synthetic purine analog, is approved for the treatment of HIV-1 infection, in combination with other antiretroviral agents. Abacavir is available in a co-formulation with zidovudine and lamivudine (TRIZIVIR) for twice-daily dosing and in a co-formulation with lamivudine (EPZICOM) for once-daily dosing. Abacavir is approved for use in adult and pediatric patients ≥3 months of age, with dosing in the latter based on body weight.
MECHANISMS OF ACTION AND RESISTANCE. Abacavir is the only approved antiretroviral that is active as a guanosine analog. It is sequentially phosphorylated in the host cell to carbovir 5′-triphosphate, which terminates the elongation of proviral DNA because it is incorporated by reverse transcriptase into nascent DNA but lacks a 3′-hydroxyl group. Clinical resistance to abacavir is associated with 4 specific substitutions: K65R, L74V, Y115F, and M184V. In combination, these substitutions can reduce susceptibility by up to 10-fold. K65R confers cross-resistance to all nucleosides except zidovudine. An alternate pathway for abacavir resistance involves mutations at codons 41, 210, and 215.
ADME. Table 59–2 summarizes the PK parameters for this agent. The presence of food does not affect the oral bioavailability of abacavir. Abacavir is neither a substrate nor an inhibitor of CYPs. Its CSF/plasma AUC ratio is ~0.3.
UNTOWARD EFFECTS. The most important adverse effect of abacavir is a unique and potentially fatal hypersensitivity syndrome characterized by fever, abdominal pain, and other GI complaints; a mild maculopapular rash; and malaise or fatigue. Respiratory complaints (cough, pharyngitis, dyspnea), musculoskeletal complaints, headache, and paresthesias are less common. The presence of concurrent fever, abdominal pain, and rash within 6 weeks of starting abacavir is diagnostic and necessitates immediate discontinuation of the drug. Abacavir must never be restarted once discontinued for hypersensitivity. The hypersensitivity syndrome (in 2-9% of patients) results from a genetically mediated immune response linked to both the HLA-B*5701 locus and the M493T allele in the heat-shock locus Hsp70-Hom. The latter gene is implicated in antigen presentation, and this haplotype is associated with aberrant tumor necrosis factor-α release after exposure of human lymphocytes to abacavir ex vivo.
PRECAUTIONS AND INTERACTIONS. Abacavir is not associated with any clinically significant pharmacokinetic drug interactions. However, a large dose of ethanol (0.7 g/kg) increased the abacavir plasma AUC by 41% and prolonged the elimination t1/2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


