Cell Lines and Organism
A number of different cell lines have been used to propagate C. trachomatis, including McCoy, HeLa 229, and BHK-21 (clone 13). According to a recent study, it seems that the recommended cell line for susceptibility testing for C. trachomatis should be the McCoy line, and Hep-2 should be used for C. pneumoniae. To render these cells more susceptible to infection, a variety of treatments have been employed, including cycloheximide, diethylaminoethyl (DEAE)-dextran, 5-iodo-2-deoxyuridine, cytochalasin B, and irradiation. However, there is general agreement that the treatment of choice is cycloheximide. Cycloheximide is used at a concentration of 0.5 to 2.0 g/mL. Each lot should be tested for potency by dose-response curve analysis because this varies, and the optimum concentration for any lot can be determined only by experimentation. An acceptable growth medium is Minimum Essential Eagle Medium (EMEM) supplemented with 2 mmol/L glutamine, 4.4% (wt/vol) sodium bicarbonate, and 10% (vol/vol) fetal bovine serum (19).
McCoy cells should undergo at least two passages in an antibiotic-free medium to ensure that all traces of antibiotic in the growth medium have been removed. Some investigators carry out 10 to 15 passages in antibiotic-free medium before using the cells for susceptibility testing (4,19). When microdilution plates are used, the cells are seeded at a concentration of 3 × 105 cells per well, and the plates are incubated for 48 to 72 hours, at the end of which time the cells should have formed a subconfluent monolayer (4).
Despite the widespread use of HeLa and McCoy cells for susceptibility studies with C. trachomatis, these cells may not provide a relevant in vitro environment for such testing. This was demonstrated in a publication by Wyrick et al. (20), who showed that the minimal inhibitory concentration (MIC) for azithromycin was substantially lower with polarized human endometrial gland epithelial cells than with similar nonpolarized cells (0.125 g/mL and 0.5 g/mL, respectively). This was later confirmed by Paul et al. (21). However, the use of such cells is still not practical for most laboratories, and further work will have to be done to determine whether the extra effort of employing such cell systems is going to yield results that are more clinically relevant.
Chlamydial organisms should undergo at least one passage in antibiotic-free tissue culture cells, and sufficiently high-titered pools should be developed so that 102 to 103 inclusion-forming units (IFU) per coverslip are achieved. Because relatively little variation in susceptibility has been seen among different clinical isolates, investigators either have used well-characterized laboratory isolates (19) or recent clinical isolates (22–25). The latter are preferable if an attempt is being made to detect whether resistance is developing in current clinical isolates.
Antimicrobial Susceptibility Testing
Some workers prefer 1-dram shell vials, as opposed to microdilution plates, because the larger surface area of the coverslips (diameters vary from 10 to 12 mm) used with the vials makes it somewhat easier to detect low numbers of inclusions. The larger area provides greater assurance that a valid end point (MIC) will be obtained. On the other hand, if one uses high-titered pools of chlamydiae so that 102 to 103 IFU per coverslip are achieved, there is no reason why microdilution plates with 96 wells cannot be used (4,26). An alternative is to use 24-well plates with wells of a 13-mm diameter, which effectively circumvents the problem of too small a surface area (27). Probably, perfectly valid results can be obtained with microdilution plates, and if larger numbers of clinical isolates are to be tested or if larger numbers of compounds are to be evaluated, then the use of microdilution plates (or plates with at least 24 wells) is the only practical alternative.
Recommended Technique: Microdilution Plate Method
Prior to inoculation, the monolayers are exposed to DEAE-dextran at a concentration of 30 g/mL for 10 to 30 minutes (4,28). Each of the 24-well plates is inoculated with an inoculum of chlamydiae that yields 5 × 103 IFU/mL. The infectious inoculum (0.1 mL) is centrifuged onto the monolayer at 1,200 g for 60 minutes at room temperature. This centrifugation step is essential to infect cells with all C. trachomatis strains other than those that cause lymphogranuloma venereum. After centrifugation, the growth medium is removed and replaced with EMEM supplemented with glucose (5 mg/L), cycloheximide (generally 1 mg/L), 3% fetal bovine serum, and serial twofold dilutions of each antibiotic to be tested (19). All tests are performed in triplicate. The plates are then incubated at 35°C in a CO2 incubator for 48 to 72 hours. Then the coverslips are removed from the wells, fixed in absolute methanol-acetone, and stained (4,28).
Detection of Inclusions
Most investigators who have done susceptibility testing of C. trachomatis have used iodine staining for the detection of inclusions (6). One of the major disadvantages of this stain is that the inclusions may not be detected if they are particularly small or aberrant in shape. Aberrantly shaped inclusions are particularly common when C. trachomatis is cultured in the presence of β-lactam antibiotics (29). Giemsa staining has also been used, but it is evident that either direct or indirect fluorescent staining of the monolayers is the most sensitive method for detecting chlamydial antigen in cell monolayers (4,19,29). Initially, fluorescent staining for susceptibility testing used an indirect fluorescent antibody technique employing a polyclonal antibody raised in a rabbit against the same serovar E strain used in the susceptibility studies (30). However, with the availability of fluorescein-conjugated monoclonal antibodies (Syva, Palo Alto, CA; Ortho Clinical Diagnostics, Raritan, NJ; or Kallestad, Chaska, MN) to C. trachomatis, direct fluorescent staining has become the method of choice for detecting chlamydial antigen in tissue culture (4,22–24,26,31–37). In using fluorescent stains, the manufacturers’ directions should be followed. When Giemsa staining and direct immunofluorescent (using the Syva monoclonal antibody) techniques were compared, it was evident that the MICs obtainable with the monoclonal antibody were about two times higher than those obtained with Giemsa staining (29). Enzyme-linked immunosorbent assays (ELISA) have also been used to detect chlamydial antigen, and these yield MIC values comparable to those obtained with the immunofluorescent method (38). A commercially available enzyme immunoassay (Chlamydiazyme; Abbott Laboratories, North Chicago, IL) has also been used to detect antigen, and the results were similar to those obtained using a genus-specific monoclonal antibody (Ortho Clinical Diagnostics) in an immunoperoxidase test (39).
The MIC is defined as the lowest concentration of antibiotic that completely inhibits inclusion formation after 48 to 72 hours of incubation, and the minimal bactericidal concentration (MBC) is defined as the lowest concentration of antibiotic that completely inhibits the development of inclusions when the cells are disrupted at 48 to 72 hours and passed into tissue culture medium that is free of antibiotics.
Other Assays
Antibiotic susceptibility testing for Chlamydia has also been performed using flow cytometry (18). In this assay, evaluation of antibiotic activity was done at the 25-hour time point, and cells were best permeabilized using the Ortho/Permeafix treatment. The mean fluorescence intensity (MFI) of cells was determined by this method after staining of chlamydial inclusions with an anti-Chlamydia fluorescent monoclonal antibody. Calculation of the inhibitory concentration 50 (IC50), defined as the antibiotic concentration required to reduce the drug-free control MFI by 50%, by flow cytometry allowed a more objective and precise evaluation of antibiotic activity than MIC (18).
Finally, an RT-PCR–based method has been developed for antimicrobial susceptibility testing of C. pneumoniae (40) and C. trachomatis (17). The results obtained in these studies were in the range previously reported using immunofluorescent staining, and the MICs obtained by RT-PCR were consistently higher (17,40). The advantage of the RT-PCR technique over ordinary PCR methods is that only viable organisms will produce RNA.
Results of Susceptibility Testing
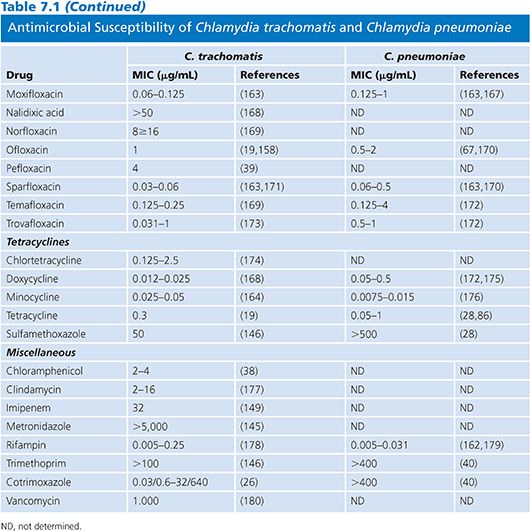
Table 7.1 presents the results of MICs of antimicrobial agents against C. trachomatis and C. pneumoniae indicating the intense interest in antimicrobials with activity against these important human pathogens. In Table 7.1, as in all subsequent tables, a range is given for the MIC values, except in those instances when so few isolates were studied that only a single MIC value is available. Aminoglycosides are without any activity and can therefore be incorporated into tissue culture media used for the isolation of this organism. β-Lactamine compounds, chloramphenicol, clindamycin, imipenem, metronidazole, and vancomycin are not active against Chlamydia. The β-lactams result in the formation of aberrant inclusions but lack significant activity. Susceptibility of C. pneumoniae is similar to that of C. trachomatis, except that C. pneumoniae is resistant to sulfonamides. The most active agents against Chlamydia are the macrolides, tetracyclines, rifampin, and fluoroquinolones. However, rifampin is not used clinically because resistance develops rapidly in vitro (41).
Antimicrobial Resistance
The development of significant resistance to currently used antimicrobials has not been a problem in human isolates, although relative resistance to sulfonamides, erythromycin, rifamycins, and fluoroquinolones has been reported for C. trachomatis (10,11,42,43), arguing for the continued surveillance of current clinical isolates. One potential explanation for the lack of resistance by chlamydiae is their unique life cycle (5). However, resistance to tetracycline in swine Chlamydia suis has been reported in the Midwestern United States (44,45). Despite the lack of evidence for frequent resistance in chlamydiae in human isolates, it is clearly possible to induce resistance in the laboratory by serial passages of organisms in subinhibitory concentrations of antimicrobials (46).
MYCOPLASMAS
Disease in humans is associated with at least four Mycoplasma species: respiratory infections with Mycoplasma pneumoniae and urogenital infections with Mycoplasma hominis, Mycoplasma genitalium, and Ureaplasma urealyticum. Finally, Mycoplasma fermentans has been isolated from patients with AIDS, and there has been conjecture about the possible role of these mycoplasmas as cofactors in the disease caused by HIV. M. fermentans has also been detected in some cases of fatal respiratory distress in immunocompetent adults (47).
The main structural characteristic of mycoplasmas is their lack of a cell wall, which makes them naturally resistant to β-lactams and all antibiotics that target the cell wall, including glycopeptides and polymyxins and agents that interfere with the synthesis of folic acid (9).
The techniques for isolating and identifying these agents are well known (48), but the techniques for performing antimicrobial susceptibility studies are less well defined (1). Both the agar dilution (49) and broth dilution (50) methods have their proponents. The two methods may yield quite disparate results for certain antibiotics, and so it is always important to consider the method used when evaluating the results of susceptibility studies (51).
A particular problem with each is that there is some drift of end points with time (a progressive increase in MIC values with prolonged incubation), as long incubation times are required for most mycoplasmas because of their slow growth. One of the probable reasons for the phenomenon of drift is that antibiotic inactivation occurs during incubation.
One advantage of the agar method is that in a mixture of sensitive and resistant strains, the two types of strains can be differentiated. With the broth method, differentiation is impossible without the cloning of isolates (49). On the other hand, from a purely clinical standpoint, it is probably of little importance to detect a mixture of resistant and susceptible strains, although it could be of considerable research interest (52).
Tanner et al. (53) adapted commercially available Sensititre broth microdilution plates for the susceptibility testing of Mycoplasma hyopneumoniae, and this technique was also successfully employed by Poulin et al. (54) for testing AIDS-associated mycoplasmas. The technique yields results comparable to those obtained with the macrodilution method.
Limb et al. (55) have utilized the measurement of ATP bioluminescence for the susceptibility testing of mycoplasmas. Using this technique, they were able to demonstrate good correlation with conventional methods and could achieve results within 6 hours.
Media and Organisms
Actively growing broth cultures are frozen at −70°C. A suitable medium for M. pneumoniae, M. fermentans, Mycoplasma incognitus, and M. genitalium is SP-4 medium (56), and a suitable medium for both of the genital mycoplasmas is 10-B broth. 10-B broth is usually made with penicillin, which should be omitted in susceptibility testing. Commercially prepared 10-B broth, as well as other specialized media for the isolation and propagation of mycoplasmas, may be obtained from commercial sources (e.g., Regional Media Laboratories, Lenexa, KS) and can be ordered without antibiotics for susceptibility testing.
For the culture of M. hominis, arginine, rather than urea, is incorporated into the medium (48). An aliquot of the culture is thawed, and serial dilutions are carried out to determine how many color changing units (CCUs) are present per milliliter. One CCU is the minimum inoculum required to produce enough growth to cause a color change in the phenol red indicator.
Antimicrobial Susceptibility Testing
The authors favor the broth dilution technique because it can be carried out in microdilution plates, allowing relatively large numbers of isolates to be tested against a reasonable number of different antimicrobials (57). Furthermore, the technique is adaptable for a variety of mycoplasmas, and when tests are carried out in triplicate, very good reproducibility is noted (57,58).
In the case of U. urealyticum, there has been good agreement noted between the more laborious tube dilution technique and the microdilution technique (59). Furthermore, both the MIC and the MBC can be determined using the broth dilution technique, whereas only the MIC can be determined using the agar dilution technique. Finally, the antibiotic broth dilution technique is really just an adaptation of the metabolic inhibition test (48), which has been used for identifying and serotyping mycoplasmas as well as for serodiagnosis. For this reason, laboratories may already be familiar with the basic components of the broth dilution technique.
The metabolic inhibition technique depends on the presence of either antibodies or, in the case of susceptibility testing, antimicrobials inhibiting the growth of the mycoplasmas. Inhibition of growth is detected by the lack of color change of a pH indicator, generally phenol red. Suitable substrates are included in the growth medium for the varying mycoplasmas: glucose in the case of M. pneumoniae, arginine for M. hominis, and urea for U. urealyticum.
Recommended Technique: Broth Dilution Technique
The broth dilution method is performed in 96-well plates with a volume of 200 µL in each well. Each stock antibiotic is added in a volume of 0.025 mL to a well of a microdilution plate, generally in triplicate (1,57). An aliquot of a previously frozen (−70°C), actively growing broth culture is thawed on the day of the assay and added to 50 mL of the appropriate broth medium (SP-4 for M. pneumoniae and 10-B broth for M. hominis and U. urealyticum) for each antibiotic to be tested. The stock culture is diluted to yield 103 to 104 organisms per microdilution well. To further establish how many CCUs have been added to each well, 10-fold dilutions of the inoculum are made to verify that at least 103 CCUs but no more than 105 CCUs have been added to each well. Inoculated broths are incubated for 2 hours at 37°C before these broths are added to the microdilution plates. Mycoplasma suspensions are added in 0.175-mL aliquots to each well containing antibiotics. The plates are sealed in plastic bags containing sterile gauze moistened with distilled water and are incubated at 35°C to 37°C under atmospheric conditions.
Three controls are included: (a) a broth control with no mycoplasmas, (b) a drug control consisting of the maximum drug concentration tested in broth alone, and (c) a mycoplasma control consisting of the mycoplasma suspension alone in a total of 0.2 mL of broth. Plates are examined after 17 to 20 hours of incubation and once daily until growth is noted in the mycoplasma control well. The MIC will be generally available for U. urealyticum at 24 hours, for M. hominis at 48 hours, and for M. pneumoniae after 5 or more days.
Determination of Minimal Inhibitory Concentration and Minimal Bactericidal Concentration
With SP-4 medium and 10-B broth, growth of U. urealyticum sufficient for determination of the MIC occurs overnight. Comparable times are 24 to 48 hours for M. hominis and 3 to 5 days for M. pneumoniae. Often, investigators determine both initial and final MICs (57); the initial MIC is the minimum amount of antibiotic required to inhibit any color change of the broth when the control well (containing organisms but no antibiotic) first shows a color change, and the final MIC is the minimum concentration of antibiotic that prevents a color change over a period of 2 consecutive days. The final MIC is employed with mycoplasmas because of their slow growth characteristics, which result in the drift of the MIC. In fact, the final MIC may be as much as eight times higher than the initial MIC for some antimicrobials (57).
It is also possible to determine the MBC, by diluting the broth from wells showing no color change in antibiotic-free medium. Generally, this is a 20-fold dilution, which is usually sufficient to dilute the antibiotic to a level below the antibiotic’s MIC value (1), but hopefully not to a point where organisms can no longer be detected. An alternate method avoids these possible pitfalls by filtering the broth from wells with no color change through a filter with a pore size of 220 nm, washing the filter free of residual antibiotic, and culturing the filter (60). Using the latter technique, Taylor-Robinson and Furr (60) were able to show that the macrolide rosaramicin acted in a purely mycoplasmastatic fashion on some ureaplasmas.
In the case of ureaplasmas, there is a self-sterilizing effect, so that by 24 hours, there is often a precipitous fall in the number of organisms (1). Because Taylor-Robinson and Furr (60) showed that there is no change in the MIC values for these organisms between 5 and 25 hours, subculturing for the determination of MBC values can be done as early as 5 hours, which avoids any problems that may be caused by the self-sterilizing phenomenon (1).
Results of Susceptibility Testing
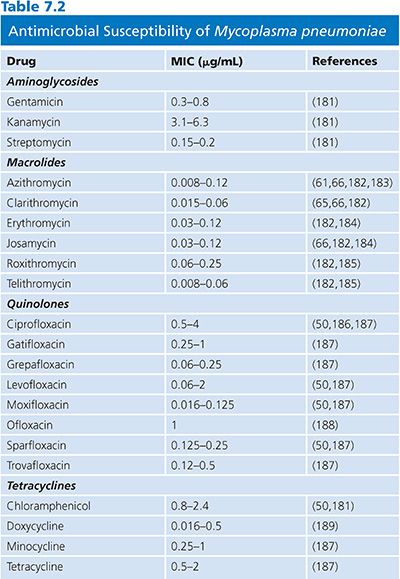
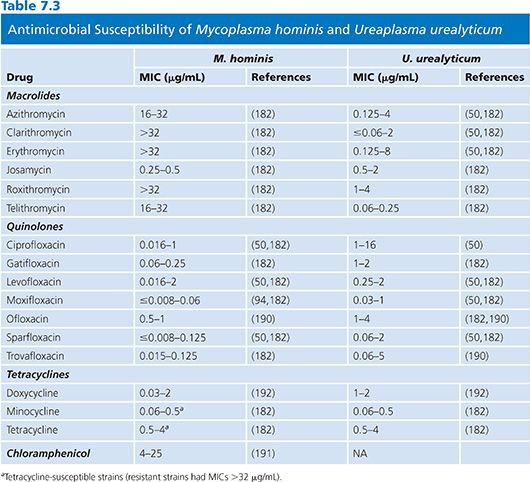
The results of susceptibility testing on mycoplasmas are summarized in Tables 7.2 and 7.3. Mycoplasmas lack peptidoglycan and penicillin-binding proteins and thus are naturally resistant to β-lactam antibiotics. Moreover, they are also resistant to rifampin, owing to the particular structure of their RNA polymerase, and also to polymyxins, nalidixic acid, sulfonamides, and trimethoprim. Tetracyclines, erythromycin, clindamycin, chloramphenicol, aminoglycosides, and fluoroquinolones have been shown to have activity against one or more mycoplasmal species (61).
Mycoplasma pneumoniae
Erythromycin and tetracyclines are usually active against M. pneumoniae. Resistance to erythromycin has now been described (62,63), although resistance to tetracycline has not been documented (64). Azithromycin and telithromycin are more active against M. pneumoniae in vitro than erythromycin, clarithromycin, or roxithromycin (32,65,66). Fluoroquinolone compounds are also active against M. pneumoniae, but in vitro studies have shown that they are not as effective as macrolides (67–69). Clinical strains with acquired resistance to macrolides have recently emerged worldwide with resistances to erythromycin and azithromycin mainly due to mutations in 23S rRNA (64).
Mycoplasma hominis
M. hominis is usually naturally susceptible to tetracyclines but tetracycline-resistant isolates containing DNA sequences homologous to the streptococcal determinant tetM have been reported (52). It has been convincingly demonstrated that tetM is not a plasmid and that it is present in both species of genital mycoplasmas. Resistance to tetracycline in vitro is associated with failure of tetracycline treatment to eradicate M. hominis (9). A recent study from Germany has shown that the prevalence of resistance to tetracyclines and fluoroquinolones has increased between 1989 and 2004, but doxycycline still remains the drug of choice for the treatment (70). The new glycylcyclines have been shown to be active in vitro against M. hominis strains resistant to other tetracyclines. Because of this emerging resistance, it is proving to be increasingly difficult to devise suitable antimicrobial regimens for the effective therapy of genital mycoplasma infections (9,71–73). Clindamycin can be used for the treatment of M. hominis infections resistant to tetracyclines, and erythromycin or quinolones can be used for tetracycline-resistant U. urealyticum infections (9,39). Usually, fluoroquinolone compounds are active against M. hominis, but resistance to fluoroquinolones has been reported and is associated with mutations in DNA gyrase (74). A fatal case of a disseminated infection with clindamycin and ciprofloxacin-resistant M. hominis has been recently reported in Germany (75). M. hominis is resistant to erythromycin, roxithromycin, azithromycin, and clarithromycin but remains susceptible to josamycin (Table 7.3).
Ureaplasma urealyticum
Tetracyclines and fluoroquinolone compounds are active against U. urealyticum. Erythromycin is generally active against ureaplasmas, but resistance has been noted (14) and is associated with specific mutations in the 23S rRNA gene (76).
RICKETTSIA
All members of the genus Rickettsia are obligate, gram-negative, intracellular bacteria. The genus comprises typhus group rickettsiae, which include Rickettsia prowazekii, the agent of epidemic typhus, and Rickettsia typhi, the agent of murine typhus; Orientia tsutsugamushi, the agent of scrub typhus (77); and spotted fever group (SFG) rickettsiae. The number of recognized SFG rickettsioses has recently increased. The six SFG rickettsioses previously described are Rocky Mountain spotted fever, caused by Rickettsia rickettsii; Mediterranean spotted fever, caused by Rickettsia conorii subsp conorii; Siberian tick typhus, caused by Rickettsia sibirica subsp sibirica; Israeli spotted fever, caused by R. conorii subsp israelensis (77); Queensland tick typhus, caused by Rickettsia australis; and rickettsialpox, caused by Rickettsia akari. Since 1984, 12 new SFG rickettsiosis have been described (77): the Japanese or Oriental spotted fever, caused by Rickettsia japonica and described in 1984; Flinders Island spotted fever, caused by Rickettsia honei and described in 1991; Astrakhan fever, caused by R. conorii subsp caspia and reported in 1991; African tick-bite fever, caused by Rickettsia africae and described in 1992; a new spotted fever due to Rickettsia mongolitimonae, reported in France in 1996; Rickettsia slovaca infection, reported in 1997; Rickettsia helvetica infection, described in 2000; flea-borne rickettsioses, caused by Rickettsia felis and reported in 2001; Rickettsia aeschlimannii infection, reported in 2001; Rickettsia heilongjiangensis infection; Rickettsia parkeri infection, reported in 2003; Rickettsia massiliae; and Rickettsia marmionii (77).
All of the members of the genus Rickettsia are obligate intracellular pathogens and, therefore, require either animal models, embryonated eggs, or tissue culture for susceptibility assays (78). It would appear that, of the in vitro techniques now available, the plaque assay and a colorimetric assay (78) are the most practical for evaluating antiinfectives for this group of organisms. However, because of the technical difficulties in working with these agents, susceptibility testing will probably be confined to relatively few laboratories. Furthermore, it is evident that when in vivo techniques, such as suppression of lethality in chicken embryos, are compared with in vitro techniques, such as the plaque assay, the results may be somewhat discrepant (79). The two assays (plaque assay and colorimetric assay) depend on the induction of cytopathic effects and plaque formation in cell cultures by the rickettsiae, but some rickettsiae do not normally cause cytopathic effects in primary cultures (80,81). Recently, Ives et al. (82,83) described a new assay that uses immunofluorescent staining, which avoids the problem of a lack of cytopathic effects. Very recently, we have developed a new quantitative PCR DNA assay using the LightCycler system for the evaluation of antibiotic susceptibilities of three rickettsial species, including R. felis, a rickettsial species that does not induce plaque in cell cultures (81).
Cell Lines and Organisms
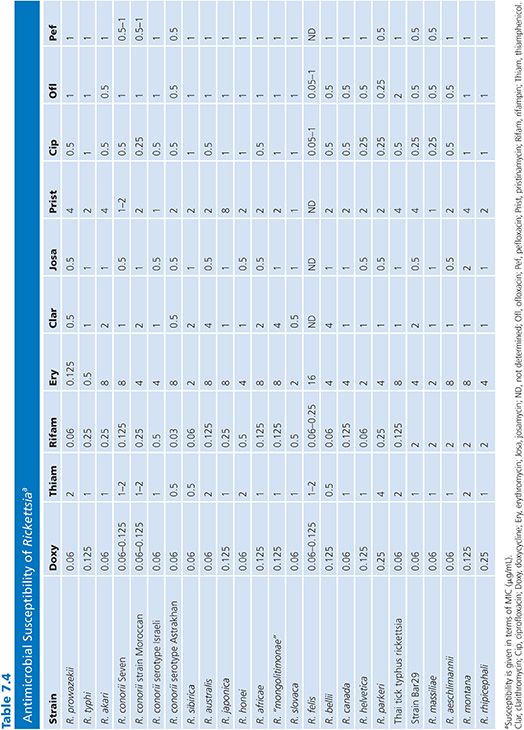
Organisms used for these studies are laboratory strains. For example, in the case of R. rickettsii, the Sheila Smith strain is used (78), and for R. conorii, the American Type Culture Collection VR 141 Moroccan strain is used (84). Only a few studies on in vitro antibiotic susceptibilities of SFG rickettsiae other than R. conorii, R. rickettsii, and R. akari are available. We recently reported an extensive study that investigated the reaction of 27 rickettsiae to 13 antimicrobials (Table 7.4) (80).
Antimicrobial Susceptibility Testing
Reference Method: Plaque Assay
Vero cell monolayers seeded 24 hours before use in round, plastic, tissue culture Petri dishes (60 mm; Corning Glass Works, Corning, NY) are infected with 1 mL of a solution containing 4 × 103 plaque-forming units (PFU) of the desired rickettsial strain (78). After 1 hour of incubation at room temperature (22°C), the plates are overlaid with 4 mL of a medium containing Minimum Essential Eagle Medium (Gibco Laboratories, Grand Island, NY), 2% newborn calf serum, 2% (N-[2-hydroxyethyl]) piperazine-N ′-[2-ethanesulfonic acid]) (HEPES, Sigma-Aldrich, St. Louis, MO), and 0.5% agar. The antibiotic solutions are added to the medium to obtain the desired final concentrations (an antibiotic-free control plate is also included), and the plates are incubated for 4 to 7 days at 35°C in a CO2 incubator. All antibiotics are assayed in triplicate, at a minimum. After incubation, the monolayers are fixed with 4% formaldehyde and stained with 1% crystal violet in 20% ethanol. The MIC is the lowest concentration of the agent tested causing complete inhibition of plaque formation, compared with the drug-free controls. Plates may then be photographed to obtain a permanent record of the results. The major problem with this technique is that some rickettsial strains may not induce the formation of plaques in cell cultures. In this case, several passages in various cell lines may allow selection of variants able to produce plaques.
Rickettsiacidal activity can be determined from this assay by staining surviving cells with either Gimenez or immunofluorescent stain (79). The minimal concentration of antibiotic that completely sterilizes the monolayer is defined as the minimum rickettsiacidal concentration.
Dye-Uptake Assay
Flat-bottomed microdilution plates are seeded with 1.5 × 104 Vero cells (suspended in a solution of EMEM, 5% newborn calf serum, and 2 mmol/L L-glutamine) per well and subsequently infected with varying concentrations of a suspension of Rickettsia organisms (78). The Vero cell suspension (100 µL) is added to each well. The infectious inoculum is added to individual wells in a final volume of 50 µL. For each 96-well plate, the first horizontal row of 8 wells contains no rickettsiae, the second horizontal row is inoculated with 2,000 PFU, the third horizontal row with 200 PFU, and the fourth with 20 PFU (Fig. 7.1). Two antibiotics are tested per plate, four concentrations of each antibiotic are tested, and each concentration of antibiotic is replicated 12 times; that is, each row contains the same antibiotic at the same concentration. Antibiotics are added in 50-µL volumes. Incubation is then carried out at 36°C for 4 days in a CO2 incubator.
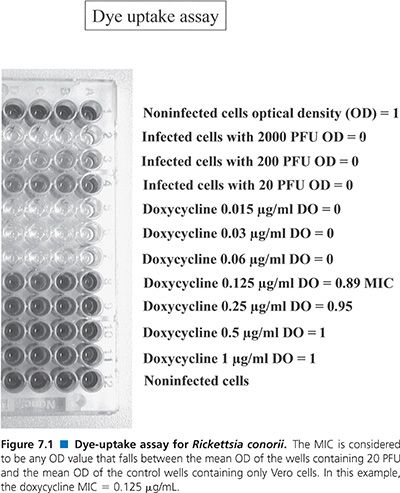
After this, the medium is removed, 50 µL of neutral red dye (0.15% in saline, pH 5.5; Sigma Chemical Co, St. Louis, MO) is added to each well, and the plate is incubated for 60 minutes at 36°C. Unincorporated dye is then washed (three washes) from the cells using phosphate-buffered saline (pH 6.5). Incorporated dye is removed from the well using 100 µL of phosphate-ethanol buffer (10% ethanol in phosphate-buffered saline, adjusted to pH 4.2).
Finally, the optical density (OD) of the solution is read at 492 nm with a multichannel spectrophotometer designed for use with microdilution plates (EIA Autoreader, model EL310; BioTek Instruments, Winooski, VT). The mean OD of the control wells (containing only Vero cells) is assigned a value of 1, and the mean OD of the wells containing 2,000 PFU is assigned a value of 0 (Fig. 7.1). The MIC is considered to be any OD value that falls between the mean OD of the wells containing 20 PFU and the mean OD of the control wells containing only Vero cells.
This assay is an adaptation of an assay used to determine the efficacy of agents against herpes viruses (85) and is dependent on the fact that intact cells take up neutral red dye. Hence, wells that contain fewer cells take up less neutral red and yield lower OD values. This test is, therefore, a derivative of the plaque assay but has the advantage of using microdilution technology as well as an automated means of reading the plates.
Other Assays
Immunofluorescence Assay. More recently, an immunofluorescence assay was described by Ives (83). In this model, Vero cells cultured in wells of chamber culture microscope slides were infected with rickettsiae. After incubation of cultures for 3 hours at 37°C in a 5% CO2 atmosphere, cell supernatants were replaced by new medium containing various concentrations of the antibiotics to be tested. Drug-free cultures served as controls. Cell culture monolayers were then fixed with methanol and stained using an immunofluorescence assay to reveal the presence of immunofluorescent foci (clusters of rickettsiae) in 25 random fields for each well. The minimal antibiotic concentration allowing complete inhibition of foci formation as compared with the drug-free controls was recorded as the MIC.
LightCycler Polymerase Chain Reaction Assay. Cells cultured in 24-well plates were infected with rickettsiae and incubated for 7 days at 37°C in a 5% CO2 atmosphere with medium containing various concentrations of the antibiotics to be tested. Wells were harvested each day for 7 days and stored at −20°C before the PCR assay. Real-time PCR was performed on LightCycler instrumentation (Roche Biochemicals, Mannheim, Germany) (86). The specificity of amplification can be confirmed by melting curve analysis. Single melting peaks can be generated by depicting the negative derivative of fluorescence versus temperature (−dF/dT) over the course of a gradual PCR product melt.
Extraction of DNA. After thawing, harvested tubes were centrifuged at 5,000 rpm for 10 minutes, supernatant was discarded, and pellet was washed twice with sterile distilled water and finally resuspended with 200 µL of sterile distilled water. Extraction of the DNA was performed using Chelex (biotechnology-grade chelating resin, Chelex 100, Bio-Rad, Richmond, CA) at 20% in sterile water. Briefly, 500 µL of Chelex was added to each tube, then the tubes were vortex-mixed and placed in a boiling water bath for 30 minutes. The tubes were then centrifuged at 14,000 rpm for 10 minutes, and supernatant was harvested and stored in sterile tubes at 4°C before use.
Polymerase Chain Reaction Master Mix. Master mixes were prepared by following the manufacturer’s instructions, using the primers CS877F (5′-GGG GGC CTG CTC ACG GCG G-3′) and CS1258R (5′-ATT GCA AAA AGT ACA GTG AAC A-3′) of the citrate synthase gene. The 20-µL sample volume in each glass capillary contained the following: for all single experiments, 2 µL of LightCycler DNA Master SYBR Green (Roche Biochemicals, Mannheim, Germany), 2.4 µL of MgCl2 at 4 mM, 1 µL of each primer at 0.5 µM, 11.6 µL of sterile distilled water, and 2 µL of DNA.
Polymerase Chain Reaction Cycling and Melting Curve Conditions. After one-pulse centrifugation to allow mixing and to drive the mix into the distal end of each tube, glass capillaries were placed in the LightCycler instrument. The amplification program included an initial denaturation step consisting of 1 cycle at 95°C for 120 seconds and 40 cycles of denaturation at 95°C for 15 seconds, annealing at 54°C for 8 seconds, and extension at 72°C for 15 seconds, with fluorescence acquisition at 54°C in single mode. Melting curve analysis was done at 45°C to 90°C (temperature transition, 20°C per second), with stepwise fluorescence acquisition by real-time measurement of fluorescence directly in the clear glass capillary tubes. Sequence-specific standard curves were generated using 10-fold serial dilutions (105 to 106 copies) of a standard bacterial concentration of Rickettsia organisms. The number of copies of each sample transcript was then calculated from a standard curve using the LightCycler software. The MIC was defined as the first antibiotic concentration allowing the inhibition of growth of bacteria as compared with the number of DNA copies at day 0. Experiments were made twice in duplicate. This technique is specific, reproducible, easy to perform, and rapid. It can also be used to measure the number of DNA copies at any time, and we were able to perform for the first time a kinetic of the growth of Rickettsia organisms even if the bacteria did not lead to plaque in vitro in cell cultures.
Results of Susceptibility Testing
The results of susceptibility testing of Rickettsia species are presented in Table 7.4. Sensitivities to amoxicillin (MICs from 128 to 256 µg/mL), gentamicin (MICs from 4 to 16 µg/mL), and co-trimoxazole were poor (80,81). Doxycycline was the most effective antibiotic against all strains tested, with MICs ranging from 0.06 to 0.25 µg/mL. The MICs of thiamphenicol ranged from 0.5 to 4 µg/mL, and the MICs for fluoroquinolone compounds ranged from 0.25 to 2 µg/mL. Among the macrolide compounds, josamycin was the most effective antibiotic, with MICs ranging from 0.5 to 1 µg/mL. Typhus group rickettsiae were susceptible to erythromycin (MICs from 0.125 to 0.5 µg/mL), whereas SFG rickettsiae were not (MICs from 2 to 8 µg/mL), and this difference is likely due to mutations in L22 ribosomal protein for SFG rickettsiae (87). Recently, we demonstrated that the new ketolide compound telithromycin was very effective against typhus group rickettsiae and SFG rickettsiae, with MICs ranging from 0.5 to 1 µg/mL (97). Susceptibilities to rifampin varied: typhus group rickettsiae and most SFG rickettsiae were susceptible (MICs from 0.03 to 1 µg/mL), but a cluster including R. massiliae, Rickettsia montana, Rickettsia rhipicephali, R. aeschlimannii, and strain Bar 29 were more resistant (MICs from 2 to 4 µg/mL). This relative resistance to rifampin was linked to natural mutations in the rpoB gene (88).
EHRLICHIA
Ehrlichioses are emerging infectious diseases caused by obligate, gram-negative, intracellular bacteria belonging to the Proteobacteria α subgroup (89). The genus Ehrlichia is divided into three genogroups: the group Neorickettsia, with Neorickettsia sennetsu, Neorickettsia risticii, and Neorickettsia helminthoeca; the group Ehrlichia, with Ehrlichia canis, Ehrlichia chaffeensis, Ehrlichia rumitantium, Ehrlichia muris, and Ehrlichia ewingii; and the group Anaplasma with Anaplasma platys, Anaplasma marginale, and Anaplasma phagocytophilum (89,90).
They are responsible for human and animal diseases. E. chaffeensis is the agent of human monocytic ehrlichiosis (HME); A. phagocytophilum, the agent of human granulocytic ehrlichiosis (HGE); and E. canis, the agent of canine ehrlichiosis. In vitro and in vivo antibiotic susceptibility studies have been carried out on various species of Ehrlichia. All have found that doxycycline and rifampin are highly effective against ehrlichiae, and thus they are currently preferred for treating animal and in human ehrlichiosis.
Antimicrobial Susceptibility Testing
Animal Models
N. sennetsu was first isolated in mice, and subsequently, infections in mice have been used as a model for Sennetsu fever. Although the growth of N. sennetsu in mice is much slower than the growth of other rickettsiae, treatment of mice with cyclophosphamide prior to inoculation has been found to enhance the growth of N. sennetsu (91), and this technique has been used for the preparation of antigen in mice. The first study of antibiotic susceptibility in mice for N. sennetsu has shown that erythromycin, sulfisoxazole, penicillin, streptomycin, polymyxin B, bacitracin, and chloramphenicol were ineffective even at high concentrations (92). Chlortetracycline was more effective than oxytetracycline and tetracycline. Further studies have evaluated the effect of tetracycline therapy on spleen size as a percentage of body weight and on the splenic infectious burden in mice infected with N. sennetsu (93,94). In mice, in which tetracycline therapy was initiated at the same time as inoculation, there were no detectable ehrlichiae in the spleen (93). Therefore, it would appear that the time of initiation of treatment may be important in controlling the course of infection with N. sennetsu and that delayed therapy may allow the development of chronic infections.
Cell Culture Model
The susceptibility of Ehrlichia species to various antibiotics has been tested using ehrlichiae-infected contact-inhibition-growth cell lines incubated for 48 to 72 hours in the antibiotic concerned. Thereafter, the antibiotic-containing media is removed, and ehrlichiae-infected cells are incubated with antibiotic-free media for at least 3 more days. The number of ehrlichiae-infected cells is counted every day, and an antibiotic is considered ineffective if the number of ehrlichiae-infected cells after exposure to the antibiotic is similar to that of noninfected control cells. If the number of ehrlichiae-infected cells is found to decrease during incubation with an antibiotic, the antibiotic is regarded as being bactericidal. Antibiotics are considered bacteriostatic if there is no increase or decrease in ehrlichiae-infected cells when the antibiotic is present but the number of infected cells increases when antibiotic-free media is provided.
Results of Susceptibility Testing
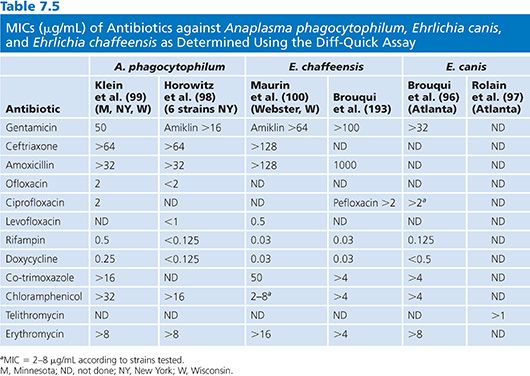
Results of susceptibility testing for A. phagocytophilum, E. canis, and E. chaffeensis using the Diff-Quick assay are presented in Table 7.5. The in vitro susceptibility of N. sennetsu Miyayama strain to eight antibiotics was determined using Diff-Quick staining of infected P388D1 cells over a 5-day period (95). In this study, it was also demonstrated that Diff-Quick staining was as reliable as immunofluorescence assay for detecting infected cells. It was found that N. sennetsu was not susceptible to penicillin, gentamicin, co-trimoxazole, erythromycin, and chloramphenicol, whereas rifampin, doxycycline, and ciprofloxacin were effective, with MICs of 0.5, 0.125, and 0.125 µg/mL, respectively.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


