FACTORS IN AGAR COMPOSITION
Antagonists of Folate Synthesis Inhibitors
Paraaminobenzoic acid (p-ABA) is a potent inhibitor of sulfonamides. Concentrations found in certain media such as peptone water and nutrient agar will virtually eliminate sulfonamide activity (26). Susceptibility testing agar used in any of the current published methods has minimal concentrations of p-ABA.
Thymidine and thymine in sufficient concentrations antagonize the dihydrofolate reductase (DHFR) inhibitors such as trimethoprim, increasing MICs and reducing zone diameters in agar diffusion tests. This nucleoside and its pyrimidine base possibly act by competing for the target enzyme. Thymidine is by far the more potent of the two, and methods to reduce or eliminate its presence in agar will restore DHFR inhibitor activity. Indeed, one common method is to use lysed horse blood, which is rich in the enzyme thymidine phosphorylase, which converts thymidine to thymine and 2-deoxyribose-1-phosphate. Further, most bacteria, except Enterococcus faecalis, cannot utilize thymine as a substrate (26). Some bacterial strains require thymidine for growth and will grow poorly or not at all on susceptibility testing agar. Because such strains are naturally DHFR inhibitor resistant, no problems of testing occur when thymidine is added back into the medium.
Although Mueller-Hinton agar is purportedly low in thymidine and thymine, occasionally, problems can arise. CLSI has developed a quality control procedure to test for low thymidine content using either of two American Type Culture Collection (ATCC) strains of E. faecalis and trimethoprim-sulfamethoxazole disks (27). This procedure is now embraced in the new ISO standard to Mueller-Hinton lot acceptance criteria (17).
Calcium, Magnesium, Zinc, and Manganese
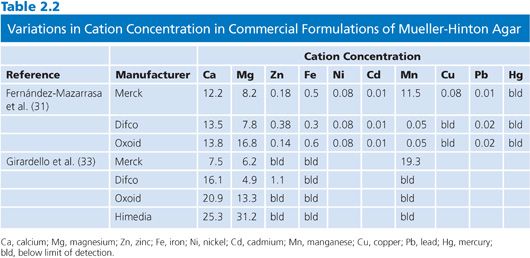
The principal problem with Mueller-Hinton over the years has been the variability in the concentrations of divalent cations, especially calcium (Ca++) and magnesium (Mg++), which can have significant effects on the activity of aminoglycosides and some other antimicrobials, particularly against Pseudomonas aeruginosa (28,29). For the broth, this has now been overcome during the manufacturing process, and the concentrations are adjusted to within an acceptable range specified by ISO 16782 (17). However, the addition of agar to Mueller-Hinton will alter the cation concentration in unpredictable ways, such that for some newer antimicrobial agents heavily influenced by cation concentration, such as Ca++ with daptomycin (30) and manganese (Mn++) with tigecycline (31,32), it has not been possible to develop agar dilution or diffusion standards. The great variability in cation concentrations with different brands of Mueller-Hinton agar has recently been highlighted (31,33).
The divalent cations of Ca++ and Mg++ are well-known antagonists of aminoglycosides. The antagonism is complex and cannot simply be accounted for by the concentrations of the cations themselves (34). It appears to be affected to a large extent by other constituents such as sodium chloride and phosphate. The effects are most obvious when testing P. aeruginosa (35–37). The variability of concentrations in Mueller-Hinton agar in the past has been one reason that certain European countries have favored Iso-Sensitest agar, where the divalent cation concentrations are defined in the formulation. Ostensibly, agar itself is processed to remove free cations and anions (14), but recent studies have demonstrated that there can still be a wide range of calcium and magnesium concentrations in different batches of Mueller-Hinton agar (30,31,33). In contrast to Mueller-Hinton broth, the concentration of divalent cations in Mueller-Hinton is not stipulated in the ISO 16782 standard. However, other susceptibility test media have not been subject to control of their divalent cations, and the reproducibility of aminoglycoside results with different lots has not been examined.
The concentration of Ca++ is critical to the interpretation of daptomycin susceptibility (30). Daptomycin activity varies greatly with Ca++ concentration, and specified concentrations are required in the media (usually those seen physiologically). For this reason, daptomycin has not yet been completely standardized for tests in agar, and supplementation is required for broth testing (30). Cation concentrations are also known to affect the activity of the polymyxins in susceptibility tests at least for P. aeruginosa and Acinetobacter baumannii (33,35). Calcium and magnesium concentrations can also affect the action of tetracycline against Pseudomonas species, although this is of little importance because tetracyclines are not considered clinically active against these species.
The concentrations of another cation, zinc (Zn++), in Mueller-Hinton (38–40) agar and Iso-Sensitest agar (41) has an impact on the activity of imipenem and possibly other carbapenems, at least for common nonfermentative gram-negative. Again, the concentrations of Zn++ can be quite variable in different brands of Mueller-Hinton agar (32,33).
A summary of two recent publications that have examined cation concentrations in Mueller-Hinton agar is presented in Table 2.2.
Sodium Chloride
Less well known is the effect of sodium chloride concentration on the activity of aminoglycosides (42,43). As summarized by Waterworth (34), variations in NaCl can have quite significant effects: an increase in concentration from 22 to 174 mM can increase the MIC of gentamicin by as much as 32-fold. NaCl is not part of the Mueller-Hinton formulation, but the manufacture to a reference standard at least generates consistent results. NaCl is part of the Iso-Sensitest formulation, but viable amounts could also come from the hydrolyzed casein and peptones.
pH
Major variation in the pH of the medium can result in major changes in the activity of aminoglycosides, macrolides, and tetracyclines. Aminoglycoside activity is substantially increased in alkaline conditions and substantially inhibited in acidic conditions. Similar effects are observed with macrolides. Susceptibility testing agars are manufactured to performance standards of pH, and in some testing methods, such as those of CLSI, it is recommended to confirm the pH in the cooled, prepoured state or with a surface pH meter after pouring, once the agar has been prepared from the dried powder in the routine laboratory.
The pH effect does become significant, however, when agar plates are incubated in increased concentrations of CO2; such is recommended for streptococci and Haemophilus species in most methods. Rosenblatt and Schoenknecht (44) have shown an increase in pH from 7.4 to 8.4 over a period of 24 hours when Mueller-Hinton blood agar plates are incubated in 5% to 7% CO2 in air. Carbon dioxide is absorbed onto the surface during incubation, some of which will be converted to carbonic acid initially and then carbonate ions, first decreasing and later increasing the pH at the surface (44). Acidity is known to reduce the activity of macrolides in particular (including the azalides and ketolides) (45–55) and of aminoglycosides to some extent (56,57) while increasing the activity of tetracyclines (2). In agar-based tests, this effect will result in higher MICs and smaller zones for macrolides and aminoglycosides, as the pH at the surface will be more acidic at the critical time.
Additives
Some bacterial species require the addition of specific nutrients to ensure adequate growth. The most common of these is blood, usually sheep or horse blood, at a concentration of 5%. When testing sulfonamides, horse blood is preferred, as it is low in sulfonamide antagonists.
Specific reagent additives are required for certain fastidious species or for nonfastidious species against certain antimicrobial agents, as described in Table 2.3.
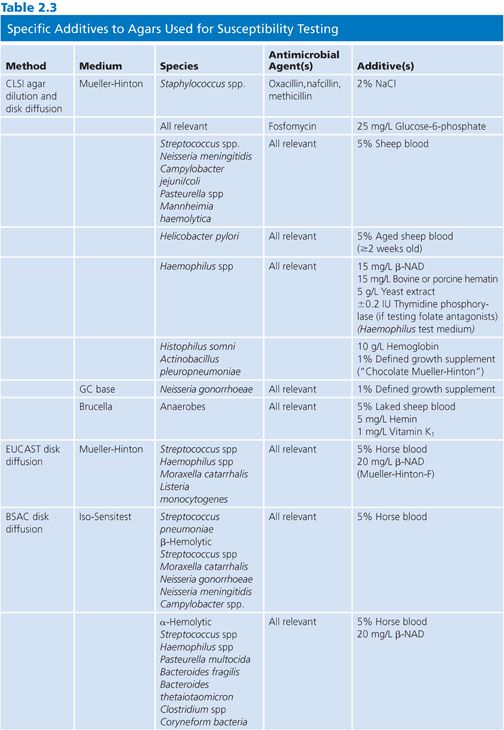
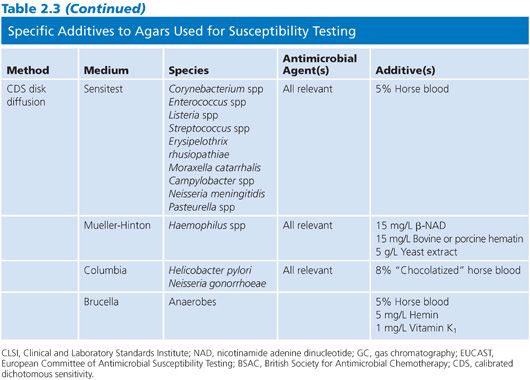
Defined Growth Supplement
One important additive, noted here and in CLSI documents as defined growth supplement, is a complex mixture of vitamins, cofactors, and other nutrient substances. The two most recognizable and important brands are IsoVitaleX (Becton, Dickinson and Company, Sparks, MD) and Vitox (Thermo Scientific, West Palm Beach, FL). These products contain 1.1 g L-cystine, 0.03 g guanine, 3 mg thiamine HCl, 13 mg p-ABA, 0.001 to 0.012 g vitamin B12, 0.1 g cocarboxylase, 0.25 g nicotinamide adenine dinucleotide (NAD), 1 g adenine, 10 g L-glutamine, 100 g glucose, 0.02 g ferric nitrate, and 25.9 mg cysteine HCl per liter of water. It is most commonly used at a 1% concentration to ensure the growth of the fastidious organisms, especially Neisseria gonorrhoeae.
PARANITROPHENYLGLYCEROL AND OTHER ANTISWARMING AGENTS
Paranitrophenylglycerol (PNPG) has been used among other techniques to prevent the swarming of Proteus mirabilis and Proteus vulgaris across agar surfaces. This is mostly a problem for agar dilution testing when multiple strains including strains of these two species are being tested on a single series of plates. As a result of the swarming, spots close to the Proteus species can be difficult or impossible to read. However, PNPG has been shown to affect the MIC results of a number of bacterial species and antimicrobials, especially Pseudomonas aeruginosa (58,59).
A second antiswarming agent, Dispersol LN, has also been evaluated and shown to affect the activity of some antimicrobials (60). The addition of PNPG or another antiswarming agent is no longer recommended routinely in any method and should only be used if there are data to show that the agent does not interfere with the MICs of that organism–antimicrobial combination. Another natural chemical, 10′(Z),13′(E)-heptadecadienylhydroquinone, has been shown to increase susceptibility of P. mirabilis to polymyxin B (61).
Higher concentrations of agar (e.g., 2%) can be used to inhibit swarming. None of the three options—use of PNPG, use of Dispersol LN, or increasing the concentration of agar—is now recommended.
Strains with Special Growth Requirements
Special problems arise with strains or species with unusual growth requirements. The so-called nutritionally variant “streptococci” Abiotrophia defectiva (previously Streptococcus defectivus) and Granulicatella adiacens (previously Streptococcus adjacens, Abiotrophia adiacens) require pyridoxal for growth. Media can be supplemented with pyridoxal (0.001%) and lysed horse blood to ensure growth of the strains (63–65) and allow interpretation of MICs at least (no breakpoints have been determined).
Occasionally, mutant strains of Staphylococcus aureus will depend on thiamine or menadione (vitamin K3) for growth. These often exhibit small colonies on primary isolation. The addition of thiamine (2 mg/L) and menadione (0.5 mg/L) to susceptibility testing media will allow susceptibility (MIC) tests to be performed (66).
Strains of Escherichia coli and other gram-negatives dependent on thymine for growth are sometimes encountered. They can be selected for during treatment with folate synthesis inhibitors (67). Strains of Enterobacteriaceae dependent for growth on thymidine, cysteine, or glutamine may be tested by adding the appropriate nutrient to the basal medium (7), although experience with this is limited due to the infrequency with which these strains are isolated.
AGAR DILUTION SUSCEPTIBILITY TESTING
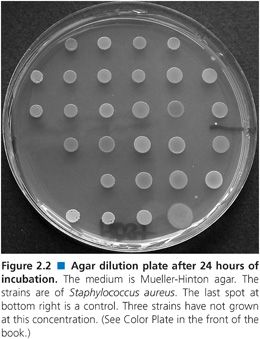
Agar dilution susceptibility testing is the solid equivalent of broth dilution susceptibility testing, either in macro- or microbroth format. One advantage it offers over broth-based methods is that it allows the simultaneous testing of a large number of strains on a single agar plate, for example, 32 on a 90-mm plate using a Steers-Foltz or similar replicator (Fig. 2.1). It is therefore well suited to the rapid evaluation of new compounds or for large-scale centralized surveillance programs. The comparative disadvantages of this method are that it includes an additional variable (agar) in the medium and that, once prepared, the plates have a limited shelf life owing to degradation of the antimicrobial.

Most important, although agar dilution susceptibility testing, such as that described by CLSI (62) and EUCAST (18), has traditionally been accepted as equivalent to broth microdilution, it is not the international (ISO) reference standard, and users of agar dilution need to be aware that there may be differences from the ISO standard for some antimicrobial agent–microorganism combinations, particularly in the light of uncontrolled cation concentrations in Mueller-Hinton agar versus Mueller-Hinton broth as specified for susceptibility testing. In the future, it will be necessary to establish equivalence to the ISO standard for new antimicrobial agents as they are developed. Methods for establishing equivalence have been described (68).
The two major published agar dilution susceptibility testing methods for common human pathogens (18,62) are essentially equivalent. In the usual approach to determining MICs on agar, the antimicrobial is incorporated into molten agar over a series of twofold dilutions, gently rotated to ensure even distribution of the antimicrobial, and then poured into Petri plates. The major elements of the test are described in the following sections.
Antimicrobial Powders
Antimicrobial powders should be obtained as “pure substance” from the manufacturer or from a reputable chemical supplier (e.g., Sigma-Aldrich Corporation). It is not appropriate to use powders found in vials for parenteral drug administration, as these may contain preservatives, surfactants, fillers, or other substances that could interfere with the antimicrobial activity, and their contents may vary legally by as much as 10% above or below the label amount. Each powder should come with an expiration date and an indication of its potency and (sometimes) water content. It is vital that all these data be taken into account before preparation of stock solutions. Data should also be available from the manufacturer or another reliable source on choice of solvent and solubility before preparing stock solutions. One easily accessible source is Table 5A in CLSI’s M100 document, which is updated yearly (69).
Powders should be stored as recommended by the manufacturer. If no instructions for storage are available, then store powders at −20°C in a desiccator, preferably under vacuum (69). This will ensure the product retains its potency for the maximum time.
Choice of Dilution Range
Before preparing stock solutions, it is essential that an appropriate dilution range be chosen. Suggested ranges for different species or bacterial types have been published (21). A full range is generally 10 to 12 doubling dilutions. Doubling dilutions are appropriate because they provide the narrowest integer series on a logarithmic scale, and MICs for a single bug–drug combination are log-normally distributed in the wild type, that is, in the absence of a resistance mechanism. Although any series could be used, the most widely accepted doubling dilution series is that of base 2. This implies that preferably the highest concentration (the concentration at the start) should be a power of 2 (e.g., 27 = 128). Following this pattern will allow comparison with the majority of published data and MICs for published quality control strains.
Preparation of Stock Solutions
Stock solutions should be prepared by weighing out the exact amount of powder using a balance designed for milligram amounts. Ideally, amounts less than 100 mg should not be weighed out. Alternatively, an approximate amount can be weighed out, and using the following formula, the exact volume can be added. The amount weighed is calculated using the formula
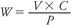
where W = weighted amount (µg), V = volume of stock solution required (mL), C = concentration of solution required (µg/mL), and P = potency in µg/mg. Usually, a potency value is provided. If not, it will need to be calculated from the values provided in the certificate of analysis. These values are purity (as measured, e.g., by high-performance liquid chromatography [HPLC]), water content (as measured, e.g., by Karl Fischer analysis), and active fraction, which will be lower for salts than for free acids or bases.
Potency = Purity × Active fraction × (1 − Water content [%])
The choice of final concentration will depend on whether the antimicrobial is being used immediately or whether it is intended for aliquoting and storage. Stock solutions for storage are best prepared as a 10-fold concentration of the highest dilution being used. Concentration choices will also be determined by the solubility of the antimicrobial. Stock solutions should be stored frozen at −20°C or lower and only thawed once before use; any unused thawed stock should be discarded.
Some antimicrobial agents require special solvents to achieve the high concentrations required in stock solutions. A comprehensive list of appropriate solvents and diluents is provided in reference 70 and is kept updated on a yearly basis as new antimicrobial agents in clinical development are added.
Preparation of Dilution Range
In order to avoid compounding minor errors in pipetting that would occur if low volumes were used or with repeated dilution, dispensing and dilution schemes such as those recommended by CLSI (69), the British Society for Antimicrobial Chemotherapy (BSAC) (21), and EUCAST (18) should be followed. These are adaptations of the original dilution scheme proposed by Ericsson and Sherris (71). An antimicrobial-free control plate should be added to any dilution series for quality control (i.e., to ensure that the selected strains are indeed viable).
Agar Selection
The description of each method identifies which agar media to use for particular bacterial groups or species. As all subsequent interpretations depend on data generated from these media, the nominated media for each method must be used. The ranges of media specified for each method are listed in Table 2.1.
Preparation of Agar Plates
Agars are generally prepared from a dehydrated base following the manufacturer’s instructions with regard to amounts of base, water, and autoclaving. After autoclaving, bottles should be cooled to 45°C to 50°C by placing them in a water bath at this temperature. When the agar has reached this temperature, antimicrobial dilutions and supplements—and blood if required—are added aseptically. An additional plate should be prepared without the incorporation of antimicrobials as a plate to ensure that there is adequate growth of test and control strains on the medium.
Adequate mixing of the antibiotic in the molten agar is essential, and this is best achieved by mixing each antibiotic solution in a small decanted volume of molten agar and then mixing the result with the bottle of molten agar. If the antibiotic solution is added directly to the whole bottle of molten agar, the solution should be warmed to avoid small portions of agar solidifying around the solution as it is added. The antimicrobial solution is mixed into the molten agar by gentle swirling and inverting the bottle rather than by shaking so that frothing is avoided. Plates should be poured onto a level surface as soon as practical after mixing.
The pH of each batch of agar should be measured after preparation. An aliquot of molten agar can be poured into a beaker or cup over a suitably designed pH electrode and allowed to gel and reach room temperature. Alternatively, a surface electrode can be placed on an aliquot of agar poured into a Petri dish and allowed to reach room temperature. It is also possible to macerate a sufficient amount of set agar and submerge the tip of an electrode.
Agar plates are then poured from the cooled molten agar as soon as possible after mixing in order to minimize any impact on the antimicrobial concentration. There is no specified depth, but 20 mL of molten agar in a 90-mm plate is considered adequate. It is advisable to dry the plates, for instance, in a fan-assisted drying cabinet for 10 minutes (21) or in a 35°C to 37°C incubator inverted with the lids off (72), as moisture buildup is common if covers are placed over cooling agar in Petri dishes.
Preparation of Inoculum
Most standards offer the choice of two methods for the preparation of inocula: the “growth” method and the “direct” method. The direct method is often preferred for inocula prepared from fastidious organisms, as growth of these organisms in broth can be a little unpredictable. The solution for final suspension varies somewhat between standards and sometimes between groups or species being tested but is generally either 0.85% to 0.9% NaCl, the same type of broth used in the agar, or phosphate-buffered saline (PBS). For reference work, inocula should be prepared from subcultures in order to ensure purity.
Calibration of Turbidity
Inocula are always calibrated to a turbidity standard, usually a McFarland turbidity standard, which employs particulate suspensions of barium sulfate. McFarland 0.5 is the most common choice and can be prepared as described in various texts (62,72). Briefly, it is a 0.5-mL aliquot of 1.175% w/v BaCl2·2H2O added to 99.5 mL of 1% w/v H2SO4 (62). Alternative turbidity standards made from other materials such as latex particles but optically equivalent to 0.5 McFarland are available commercially (e.g., Remel Inc, Lenexa, KS). Normally, a visual comparison is made with the turbidity standard. It is important that this be done in good lighting and against a card with a white background and a contrasting black line. More recently, there has been a move to the use of a nephelometer or photometer to achieve this calibration. For instance, at a wavelength of 550 nm, a 5-mL glass tube with 2 mL of inoculum suspension with an optical density of 0.1 to 0.12 approximates a 0.5 McFarland barium sulfate standard (72). Equally, at 625 nm and a light path of 1 cm, the standard has an optical density of 0.08 to 0.10 (62).
Barium sulfate standards are affected by light and heat, and hence they should be stored between uses in a dark place at room temperature. Their turbidity should be checked in a nephelometer or photometer monthly. They also require vigorous agitation at each use.
Growth Method
Usually, two to five morphologically similar colonies are picked from a primary or subculture plate. The wire loop is used to touch each colony, and it is then immersed in about 5 mL of the recommended broth (e.g., tryptic soy broth in the CLSI method). The broth is incubated at 35°C until it equals or exceeds the correct turbidity, generally 2 to 6 hours for rapidly growing pathogens. The broth is then diluted with a suitable sterile fluid such as broth, saline, or PBS according to the instructions until 0.5 McFarland is reached.
Direct Colony Suspension Method
In this method, two to five colonies are touched or picked up and suspended directly in the fluid of choice. Clearly, the turbidity should be adjusted subsequently as for the growth method. This method is acceptable in almost all situations. Hence, it remains an option. The choice of growth versus direct method depends on laboratory workflow. Almost all standards prefer the direct suspension method for fastidious organisms such as Haemophilus species and N. gonorrhoeae.
Plate Inoculation
In almost all circumstances, it is preferable to use a replicator apparatus to inoculate the agar plates, such as a Steers replicator (see Fig. 2.1). These are expensive items but last indefinitely. They generally come with 32 to 36 pins to deliver this number of strains to a 90-mm agar plate. The size of the pins is critical to decisions about how or whether the inoculum to be used for various species is to be further diluted. Prongs of 3 mm deliver approximately 2 µL (range 1 to 3 µL) to the plate surface; prongs of 2.5 mm generally deliver around 1 µL (18), while prongs of 1 mm deliver around 10-fold less (0.1 to 0.2 µL) (73). Hence, some standards recommend dilution for some organism groups or species when using the 3-mm pins.
There is some variation between standards regarding whether further dilution of the standardized inoculum prepared as outlined earlier should be performed and how it should be performed. The commonly stated intention is that each spot on the plate should contain around 104 colony-forming units (CFUs). However, in preparing standardized inocula, there are considerable differences between species in the number of CFU per milliliter for a given turbidity (74).
Incubation
Temperature and Duration
Most organisms should be incubated at 35°C to 37°C. An incubator set at 36°C can generally operate within this range. The CLSI method specifies 35°C ± 2°C for all bacteria. The duration of incubation depends on the species. For rapidly growing species such as the Enterobacteriaceae, the glucose-nonfermenting gram-negative bacilli, Enterococcus species, and Staphylococcus species, overnight incubation for a minimum of 16 hours is needed. Longer incubation is recommended for fastidious species (18 to 20 hours plus), for Staphylococcus species when testing against antistaphylococcal penicillins (24 hours), for Campylobacter species (24 to 48 hours depending on the standard and incubation temperature), for Helicobacter pylori (CLSI 3 days), and for anaerobes (42 to 48 hours).
Atmosphere
Enrichment of air with CO2 is recommended in most standards for a variety of fastidious species, including Streptococcus pneumoniae, Haemophilus influenzae, sometimes other Streptococcus species, and N. gonorrhoeae. The concentration is usually 5%, although some standards tolerate ranges of 4% to 6%. The effect on the activity of certain drugs under these circumstances was discussed earlier.
Campylobacter species and H. pylori demand incubation in a microaerophilic atmosphere, identical to that recommended for primary isolation. Commercial systems are available for generating the required microaerophilic environment.
Anaerobic bacteria should obviously be incubated in the standard anaerobic environment used for primary isolation of anaerobes. Either an anaerobic chamber or jar is acceptable.
Plate Stacking
The effect of stacking plates on the incubation temperature is not widely recognized. After placement in the incubator, plates in the middle of a stack will take longer to reach the desired incubation temperature than plates at the top and bottom (75,76). This is important because the incubation temperature can have a profound effect on the generation time of bacteria, which in turn will affect the end point determination. In general, no more than five plates should be used in a stack. Even then, it can take up to 4 hours for the center plate to reach the incubator temperature (76).
Reading
Plates should be read with optimum lighting, preferably on a dark, nonreflecting surface. For instance, in Figure 2.2, growth has obviously occurred at some spots but not at others. The MIC is taken as the first concentration at which no growth occurs. Most standards recommend that the appearance of one or two colonies or a faint haze can be ignored. However, if there are one or two colonies at a number of concentrations rather than a single one above the putative MIC, then the MIC is that concentration at which no colonies are seen. If this is the case, the purity of the strain should be checked, as it may be an indication of contamination. The only significant exception to these reading rules occurs in the reading of the end points for folate synthesis inhibitors, where growth can diminish gradually over a range of concentrations. CLSI recommends that the end point be read as that concentration resulting in 80% inhibition of growth (62). Other bacteriostatic drugs, such as chloramphenicol, clindamycin, tetracycline, and linezolid, can exhibit the same phenomenon to a lesser degree.
Quality Control
Quality control procedures in the performance of agar dilution are designed to ensure the reproducibility of the test and to confirm the performance of the reagents and the persons conducting the test. Most importantly, it is designed to detect errors of concentration or dilution of the antimicrobials, a not infrequent hazard in agar dilution testing. It is assumed that the laboratory is using reagents and materials from trusted suppliers who undertake quality control checks during manufacture. Using a trusted supplier does not prevent problems that may develop during shipping and handling, and hence, there is a need to have a quality control system at the laboratory level.
Quality Control Strains
The main component of quality control is the testing of reference quality control strains. The number tested varies between standards and with the type of strains being tested. Wherever possible, it is desirable to test a quality control strain of the same family, genus, and species as the strains under examination. The following ATCC reference strains have become almost universal as quality control strains for testing rapidly growing aerobic bacteria: Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 29213, P. aeruginosa ATCC 27853, and Enterococcus faecalis ATCC 29212. Other strains recommended by different standards include Escherichia coli ATCC 35218 (CLSI for β-lactamase inhibitor combinations), Staphylococcus aureus Collection de l’Institut Pasteur (CIP) 6525 (Comité de l’Antibiogramme de la Société Française de Microbiologie [CA-SFM] for methicillin and oxacillin), Haemophilus influenzae ATCC 49217 (CLSI and BSAC), Haemophilus influenzae ATCC 49766 (CLSI), Haemophilus influenzae National Collection of Type Cultures (NCTC) 8468 (EUCAST), N. gonorrhoeae ATCC 49226 (CLSI and BSAC), Streptococcus pneumoniae ATCC 49619 (CLSI and BSAC), Helicobacter pylori ATCC 43504 (CLSI), Campylobacter jejuni ATCC 33560 (CLSI), Bacteroides fragilis ATCC 25285 (CLSI), B. fragilis NCTC 9343 (BSAC), Bacteroides thetaiotaomicron ATCC 29741 (CLSI), Eubacterium lentum ATCC 43005 (CLSI), and Clostridium difficile ATCC 700057. The CLSI also has special control strains for veterinary testing: Histophilus somni ATCC 700025 and Actinobacillus pleuropneumoniae ATCC 27090.
The selection of quality control strains involves a compromise between the objective of getting strains with MICs close to the center of a dilution series of a range of antimicrobials (62) and the number of strains that would be required to achieve this objective. The strains commonly recommended have also been selected for their stability over long periods of time and repeated subculture. There is nothing to prevent a laboratory from developing and using its own quality control strains.
Storage of Quality Control Strains
Stock cultures of the quality control strains should be maintained in the freezer below −20°C (ideally at −70°C) in a suitable stabilizer such as fetal calf serum, glycerol broth, or skim milk or should be freeze-dried. Some workers recommend that two sets of stock cultures be stored: one set to create working cultures and the other as emergency backup. This will guarantee that the laboratory always has unaltered quality control strains. Working cultures should be stored on agar slants at 2°C to 8°C and subcultured each week for no more than 3 successive weeks. New working cultures are generated each month from frozen or freeze-dried stock and subcultured twice before use.
Frequency of Testing and Corrective Action
Each standard provides MIC quality control ranges for these strains against some or all of the antimicrobials of interest. The recommended quality control strains are included in each test run. When these strains fall within the quality control range, the test run is valid. When one or more results are out of this range, the test run must be considered invalid and be rerun.
Use in Routine Susceptibility Testing
Agar dilution can be adapted to routine susceptibility testing. Essentially, it is a truncated method that incorporates one or two selected concentrations of antimicrobial, usually at breakpoint values (so-called breakpoint susceptibility testing). It has been advocated as an option for routine testing by some authorities in the past (77) but is slowly being supplanted by other methods. The advantages and disadvantages of this method have been described in detail by BSAC (77). When testing multiple strains of gram-negative bacteria that may include P. mirabilis and P. vulgaris, it is necessary to include an antiswarming agent, which, as discussed earlier, can affect results. The method offers specific challenges in terms of quality control because the limited number of concentrations will only detect the most gross of preparation errors in standard quality control organisms and will often fail to detect the most common error—a 10-fold error in antibiotic dilutions. To overcome this, there are a number of options available: (a) assaying antibiotic dilutions prepared from stock solutions, (b) assaying agar plugs removed from a poured agar plate (78), and (c) assaying paper disks applied for a fixed interval to the surface of a poured agar plate (79). McDermott et al. (79) provided a detailed analysis of two of these methods and recommended methods with high precision.
DISK DIFFUSION SUSCEPTIBILITY TESTING
Disk diffusion susceptibility testing has a long history, having evolved out of antibiotic diffusion from wells used for drug assay and susceptibility testing. The method in its various forms still has wide popularity owing to its ease of use and low cost compared with other methods. It has spawned many variants around the world. Unlike in dilution methods, an MIC value is not generated. Instead, in the development of the test, zone diameters must be compared with the MIC values of the same strains in order to determine which zone diameters predict which MIC values and hence which category of susceptibility.
Theoretical Aspects
All disk diffusion methods are based on the diffusion through agar of drug released from an impregnated disk. There are a large number of variables affecting this diffusion. Important features of antibiotic diffusion were worked out by Cooper and others in the 1950s (75,76,80–83). They have been clearly explained by Barry (3,84), who detailed the dynamics of zone formation and the “critical concentration,” “critical time,” and “critical population.”
When an antibiotic is placed in a well cut into the agar or in a disk applied to the agar surface, the drug commences diffusion immediately and diffuses in a decreasing gradient of concentration from the edge of the well or disk. Over a number of hours, the height of this gradient deceases from very steep initially to quite shallow as the drug continues to diffuse (85). In disk susceptibility testing, disks are applied after the surface has been inoculated with bacteria. The formation of the zone edge is thus a contest between the diffusion of the drug and the growth rate of the bacterial inoculum, including any initial lag phase after incubation commences. The critical concentration is the concentration just capable of inhibiting growth, and it is also that concentration found at the zone edge at the critical time. It is similar but not identical to the MIC as measured by dilution methods. The critical time is the time it takes for the critical concentration to be reached at what ultimately becomes the zone edge. It is generally around 3 to 4 hours under standard test conditions. The critical population is the number of bacterial cells found at the critical time at the ultimate zone edge. The relationships between these parameters are as follows:
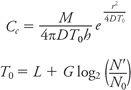
where CC = critical concentration, M = disk content, T0 = critical time, D = diffusion coefficient of drug, h = depth of agar, r = zone radius, L = lag time, G = generation time, N′2 = critical population at the critical time, and N0 = number of viable cells at beginning of incubation. Although the following description is inaccurate, it is useful to simplify these relationships by imagining the zone of inhibition as a “cylinder” in which the drug is evenly distributed. The concentration of drug in this cylinder is thus the disk content (M) divided by the volume of the cylinder, namely pd2h/4, where d is the zone diameter and h is the depth of the agar and therefore 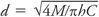 . Thus, the zone diameter increases in proportion to the square root of the disk content and decreases in proportion to the square root of the agar depth. This defines the essential relationships between zone size, disk content, and agar depth and how variations in these affect zone diameters.
. Thus, the zone diameter increases in proportion to the square root of the disk content and decreases in proportion to the square root of the agar depth. This defines the essential relationships between zone size, disk content, and agar depth and how variations in these affect zone diameters.
Disk Production
Disks for almost all antimicrobials can be obtained commercially. Even drugs that are still under development are likely to have disks available for use in the laboratory and clinical development programs. From time to time, it may be useful for a laboratory to manufacture its own disks. Strict standards must be adhered to if this is done. The same stipulations in drug sourcing and preparation of stocks as have been described in the section on agar dilution apply. Paper disks should be obtained that adhere to the same standards that apply to commercial manufacturers. In the United States, the standard is 740-E (Schleicher and Schuell, Keene, NH), and the paper used should be 30 ± 4 mg/cm2 (86).
Solvents used in disk manufacture are described in a previous edition of this book (86).
Factors Influencing Zone Diameters
A large range of factors can influence the zone sizes produced. The most important are the disk content (also called potency, mass, or strength, namely, the amount of drug in the disk), the disk size, the diffusion characteristics of the drug, the depth of the agar, the growth rate of the bacterium (including the initial lag phase), the density of the inoculum, and the activity of the drug against the strain being tested. Other factors such as medium composition, pH, and the effect of additives are dealt with at the beginning of this chapter.
Disk Content
The amount of drug impregnated into the disk is somewhat arbitrary. Amounts are chosen that are likely to produce zones of moderate size (15 to 35 mm) under normal conditions. Different disk methods have often selected different disk contents based on early experience with the antibiotic during development and precedents set within antibiotic classes.
Disk contents will be subject to variation during manufacture, and regulators such as the U.S. Food and Drug Administration (FDA) have set tolerances on the true amount in the disk in the range of 90% to 125% of the label. Such small errors will have a small effect on the zone diameter because it is proportional to the square root of the disk content, which means that the possible variation in the diameter ranges from about −5% to +12%.
The difference in zone diameters with different disk contents has been exploited to determine critical concentrations (84). When three or more disks with a range of antibiotic contents are used, the resulting zone diameters, when squared, are directly proportional to the logarithms of the disk contents. The results can be plotted using linear regression, extrapolation of which to no zone yields the critical concentrations. These values will often be good approximations of the MICs as measured by other methods (3).
Disk Size
The extent of drug diffusion will obviously be affected by the width of the disk. Paper disks are now almost universally manufactured to be 6 mm wide. Further, the nature of the paper is subject to regulation, as different varieties of paper have been shown to affect the release characteristics of antibiotics (86). One disk method, that of Neo-Sensitabs produced by the Danish company Rosco Diagnostica A/S, Taastrup, Denmark (87,88), uses 9-mm disks made from hardened inert “chalk-like” substances. These larger disks result in larger zones for the same disk content.
Diffusion Characteristics of the Drug
The two crucial properties of the drug molecules are size and charge. In general, larger molecules diffuse more slowly, and the zones formed as a consequence are smaller. Large molecules demonstrating this include the glycopeptides such as vancomycin and teicoplanin (89). Strongly cationic molecules such as the polymyxins polymyxin B and colistin will also be inhibited in their diffusion owing to interaction with acid or sulfate groups on the agar polymer. Aminoglycosides are cationic to a lesser extent, and their diffusion is reduced slightly as a result (2).
Agar Depth
As noted, agar depth will naturally alter the size of the inhibition zone. Most methods have settled on a depth of 4 mm or a similar amount. This represents a balance between a smaller depth, which is likely to generate a reasonable zone size, and a larger depth, which is designed to reduce the zone size variation due to small variations in depth. A number of studies have demonstrated greater plate-to-plate variation in zone diameter when the agar depth is less than 3 mm (89a,89b).
Time between Inoculation and Disk Placement
The period of delay between the inoculation of the agar surface and the placement of the disks prior to incubation will have a significant effect on the ultimate zone diameter. This is a direct result of the concept of “the critical time” discussed earlier. Many bacteria are capable of initiating growth at room temperature, the temperature at which plates are usually inoculated. If the plates are preincubated at 35°C, the effect will be exaggerated. Studies examining this phenomenon have been used to elucidate the critical time (3). With two exceptions, disk diffusion methods recommend that the disks be applied within 15 minutes of plate inoculation.
Incubation Time
With rapidly growing bacteria, the zone diameter is formed within a few hours of commencing incubation (3). In theory, therefore, it is possible to read zone diameters when growth becomes visible. With further incubation—recommended for all organisms in all methods—there can be subtle changes in the zone diameter beyond the time when growth first becomes visible. These changes are the result of (a) delayed growth, (b) better visualization of partial inhibition, and/or (c) the delayed appearance of resistant variants. As zone diameter breakpoints are applied to species after specified incubation periods, it is not generally possible to use these values to determine susceptibility earlier. However, if the zone of inhibition is clearly in the resistant range, further incubation is only likely to make the zone smaller, and, therefore, it would be possible to categorize a strain as resistant when growth becomes visible. Incubation of the plate for a few hours longer than typically recommended (e.g., Enterobacteriaceae for 24 rather than 18 hours) will not usually influence the interpretation significantly. Some bacteria need longer intervals of incubation than 24 hours, such as H. pylori (3 days) and some strains of Yersinia pestis (48 hours).
A specific incubation duration of 24 hours is recommended in many methods for staphylococci when tested against the antistaphylococcal penicillins, usually represented by oxacillin or methicillin, and the glycopeptides, represented by vancomycin. In parallel with broth-based susceptibility testing methods, which can use 2% NaCl, this duration is used in order to maximize the expression of strains with heteroresistance. It must be used in consort with an incubation temperature of 35°C (and no greater). In the BSAC disk method, incubation at 30°C and the addition of 2% NaCl are also used, as these factors are known to enhance heteroresistance expression (90,91). Incubation for 24 hours is also recommended for enterococci when testing them against vancomycin in the CLSI method (27).
Incubation Temperature
Incubation temperatures are designed to optimize the growth of the organisms under test. Most human bacterial pathogens are adapted to optimum growth at 37°C. Most methods therefore recommend growth at 35°C to 37°C. Incubation at 30°C is known to enhance the expression of heteroresistance to methicillin and other antistaphylococcal penicillins (90) and has been recommended as part of the recently developed BSAC disk susceptibility test (12).
Inoculum Density
Inoculum density probably has a greater effect on the ultimate zone diameter than any other variable. As described by Barry (84), the critical population is one of the three critical parameters in determining zone size. Higher inoculum densities will result in bacterial numbers reaching the critical population sooner, at a time when the critical concentration has diffused less than with lower inoculum densities (84). In the case of very dense inocula, it is likely that no zone at all will be formed, even if the organism is susceptible. Inoculum density is particularly important when the bacteria produce inactivating enzymes such as β-lactamases, especially if the enzyme requires induction, as in the case of staphylococcal penicillinase. At low inoculum densities, the drug has the ability to kill the organism before sufficient enzyme is produced, and much larger zones will result.
In the development of different methods internationally, two approaches have been taken to the choice of inoculum density. Some methods have opted for simplicity and use inocula calibrated to a turbidity standard, almost always a McFarland 0.5 barium sulfate standard. This has been shown to result in quite different inoculum densities (in terms of CFUs per milliliter) for different species (74). Denser inocula (i.e., those with higher turbidity) are required for selected species such as H. pylori to ensure prompt and adequate growth (65).
Zone Edge
The features of the zone edge will be determined by the drug activity–species relationship, the resistance mechanism (if present), and occasional peculiarities of the species. Certain drug classes are notorious for producing “fuzzy” zone edges, where there is a gradual decrease in inhibition over a distance of 1 to 5 mm. This is a standard feature of sulfonamides and will be seen sometimes with DHFR inhibitors, amphenicols, and oxazolidinones. Most methods recommend that when a fuzzy zone is seen, the reader should make a judgment as to where approximately 80% inhibition occurs and define that as the zone edge. Judging 80% inhibition accurately and consistently takes practice. Studies presented at CLSI meetings have shown that linezolid zone diameters are most reproducibly read when viewed in transmitted rather than reflected light (27).
A second phenomenon, a “heaped” zone edge, is seen occasionally, but it is a normal feature of penicillinase-producing S. aureus. It is recommended that a heaped edge be interpreted as showing that the strain is producing penicillinase.
P. mirabilis and P. vulgaris typically swarm up the disk even when susceptible to the drug. When the organism is susceptible, a thin film is observed inside a more or less distinct zone of inhibition. This film should be ignored when making zone diameter interpretations.
Disk Spacing
Although inappropriate disk spacing is a common problem, it has not been studied in any systematic way. The distance between individual disks will be determined principally by the disk contents and organism growth rate. Higher disk contents and slower growth produce larger zones. If zones are large enough, ones from adjacent disks can run into each other and reduce the chance of accurately determining the zone diameter. The CLSI method, among others, specifies the distance between adjacent disks (24 mm from center to center) to minimize the chances of zone overlap. This limits to five the number of disks that can be placed on a 90- to100-mm plate, a considerable inconvenience if six to eight drugs need to be tested, as the cost of large plates is often substantially more that the cost of the common 90- or100-mm Petri plate. The EUCAST disk method has similar recommendations, namely 6 disks for a 100-mm plate and 12 disks for a 150-mm plate (10). The additional disk for the smaller plates is possible as a number of disks in the EUCAST have strengths lower than that of CLSI. All methods agree that disks should be placed to minimize the risk of zone overlap and possible misreading.
Current Disk Susceptibility Testing Methods
Currently, there are six methods published internationally and are kept up to date through constant revision and the addition of new agents (CLSI, EUCAST, BSAC, CA-SFM, calibrated dichotomous sensitivity [CDS], and Neo-Sensitabs [Rosco Diagnostica A/S, Taastrup, Denmark]). Of these, the most widely applied are the CLSI and EUCAST methods. The other methods are largely confined in their use to single countries—BSAC in the United Kingdom, CA-SFM in France, CDS in Australia, and Neo-Sensitabs in Denmark. Methodologically, there is nothing to recommend one method over another. However, the CLSI and EUCAST processes of setting MIC breakpoints and zone diameter criteria is based on the most comprehensive data set, which includes pharmacodynamic considerations and a large amount of clinical outcome data when available. Further, these two methods are calibrated back to the now firmly established ISO reference broth microdilution method, whereas the others have not. Because zone diameter interpretive criteria are standardized for each individual method, it is not possible to either deviate from the method or use zone diameter criteria from another method unless all testing procedures and conditions are the same.
Testing of Problematic Species
Anaerobes
Two disk diffusion methods are described for anaerobes: that of the CA-SFM method (92), which uses Wilkins-Chalgren agar, and that of BSAC (12), which uses Iso-Sensitest agar supplemented with 5% horse blood and 20 mg/L of NAD. There have been several significant attempts to develop such methods in the United States (93–95), but the results have never been considered sufficiently robust for full development. In part, this relates to the prevailing view that routine testing of anaerobic bacteria is not required. Rather, it is believed that these bacteria should be tested in specific clinical circumstances or in batches to determine trends in resistance (22). BSAC has recently provided disk susceptibility testing methods for rapidly growing anaerobes on Wilkins-Chalgren agar supplemented with 5% horse blood, including interpretative criteria for a limited number of drugs (96). This effort must be considered tentative.
Campylobacter Species
After a considerable amount of work by the veterinary subcommittee of CLSI, there is now an established method for disk susceptibility testing of the principal two species (C. jejuni and Campylobacter coli) (65). The medium employed is Mueller-Hinton supplemented with 5% sheep blood. BSAC has also recently published a method using Iso-Sensitest supplemented with 5% horse blood for these two species (12). EUCAST have also recently provided a standardized disk diffusion method for these species using Mueller-Hinton-F medium (10).
Helicobacter pylori
Some authorities believe that disk susceptibility testing of H. pylori is not feasible (12,96). Their doubts are understandable given the slow-growing nature and special growth requirement of this species. However, CA-SFM has standardized a disk test on Mueller-Hinton supplemented with 10% defibrinated horse blood and developed interpretive criteria for erythromycin and ciprofloxacin (92). There have also been several preliminary studies looking at the action of macrolides and other agents against this species (97–102). There is generally excellent agreement between disk and MIC results in these methods, and testing for resistance to macrolides and metronidazole by disk only requires multilaboratory comparison and quality control development to become a fully validated method, as clinical correlates are well established (100).
Quality Control
The main features of quality control in disk susceptibility testing revolve around the regular testing of quality control strains. Each methodology has a slightly different approach and range of quality control strains. Many advocate the testing of β-lactamase–producing strains of E. coli and H. influenzae as control organisms for β-lactamase inhibitor combinations. Careful attention must be paid to the storage of quality control strains (see the earlier discussion on agar dilution). Suitable detailed guidance is provided by CLSI (27). Quality control limits are defined for the common antimicrobials and relevant control strains. Ideally, quality control tests are conducted every time that the susceptibility tests are performed. The two major methods, CLSI and EUCAST, allow the shift to weekly testing if daily testing has proven satisfactory. Usually 1 in 20 or 3 in 30 results out of range can be tolerated. Better still is the option of graphing daily results on a Shewhart chart or similar quality control chart to detect obvious trends. If there is no obvious cause for out-of-range testing, the suspect combination should be tested daily for 5 days. If all the results are within the acceptable range, normal quality control procedures can be resumed. Otherwise, a detailed analysis of the source of errors should be undertaken. Sources of error include (a) incorrect measurement or transcription of zone diameters; (b) inadequately mixed, expired, or incorrectly stored turbidity standard; (c) one or more of the materials out of date or incorrectly stored; (d) incorrect incubation temperature or atmosphere; (e) malfunctioning equipment (e.g., dispensers); (f) incorrectly stored disks; (g) altered or contaminated control strain; (h) incorrectly prepared inocula; and (i) inoculum source more than 24 hours old. Judgment is required when deciding whether patient results relevant to the out-of-range quality control tests should be reported.
Reading of Zone Diameters
Manual Methods
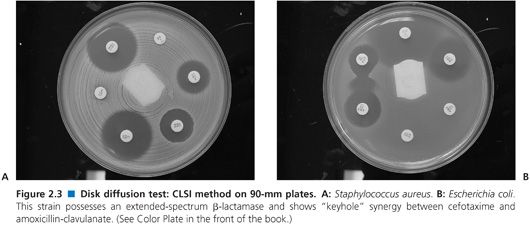
Good lighting is essential to proper manual reading of zone diameters. Optimally, the plate is held a few centimeters above a black, nonreflecting background illuminated with reflected light. For transparent media, zones should be measured from the back of the plate; for opaque media such as those containing blood, zones are measured on the upper agar surface in reflected light. Two typical plates are shown in Figure 2.3. All methods normally recommend that zone diameters be measured and recorded. Although measurement is time consuming, there are long-term advantages, as interpretive criteria can change, and retrospective adjustment of interpretation is possible when breakpoints change (as they do from time to time when new resistance emerges). Measuring and recording zone diameters are probably less frequently practiced (103). Most zone diameters are simple to read. The important exceptions are where the zone edge is not sharp, as noted earlier. Very faint growth inside the zone can be ignored. When there are multiple discrete colonies within the zone, either the strain has resistant mutants (and should be reported as resistant) or the inoculum was mixed. Either way, these colonies should be subcultured and the test repeated from the original plate or inoculum. Care should be taken with hemolytic streptococci not to read the zone of hemolysis rather than the zone edge.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


