POLYENES
Amphotericin B (AmB), natamycin, and nystatin are the currently available polyenes, although differing safety profiles have limited natamycin and nystatin to topical use (2). Polyenes possess a large lactone ring, with a lipophilic chain containing three to seven double bonds and a flexible hydrophilic portion bearing several hydroxyl groups. AmB contains seven conjugated double bonds and may be inactivated by heat, light, and extremes of pH (3). Additionally, AmB is poorly soluble and is not absorbed following oral or intramusclar delivery.
The polyenes bind to ergosterol present within the fungal cell wall membrane. This process disrupts cell wall permeability with the subsequent efflux of potassium and intracellular molecules, causing fungal death due to osmotic instability (4). There is also evidence that AmB acts as a proinflammatory agent and further serves to stimulate innate host immunity. This process involves the interaction of AmB with toll-like receptor 2 (TLR-2), the CD14 receptor, and by stimulating the release of cytokines, chemokines, and other immunologic mediators (5). It has been suggested that AmB may interact with host humoral immunity following the observation of synergistic activity of AmB and antibodies directed at heat shock protein 90 (hsp90), although further confirmatory data is needed (4). These secondary mechanims serve to increase the cascade of oxidative reactions, allowing for the rapid fungicidal activity of AmB formulations.
The spectrum of activity of AmB is broad and includes most strains of Candida, Cryptococcus neoformans, Aspergillus spp, the Mucorales, Blastomyces dermatitidis, Coccidioides spp, Histoplasma capsulatum, and Paracoccidioides brasiliensis. Candida lusitaniae, Aspergillus terreus, Fusarium spp, Pseudallescheria boydii, Scedosporium spp, and Trichosporon asahii and certain dematiaceous fungi may be resistant to AmB (6–9).
When AmB resistance occurs, it is generally attributed to reductions in ergosterol biosynthesis or the synthesis of alternative sterols with a reduced affinity for AMB. However, decreased susceptibility to oxidative damage secondary to increased catalase activity has also been described and may play a secondary role in polyene resistance (10).
Conventional amphotericin B deoxycholate (AmB-d) has been the mainstay of antifungal therapy prior to the development of other antifungal classes; yet the nephrotoxicity of AmB-d was one of the predominant forces driving the pursuit of alternative agents with a diminished side effect profile. Lipid formulations of AmB have subsequently been developed and in most instances have replaced AmB-d (11). The lipid-based formulations of AmB include amphotericin B lipid complex (ABLC), liposomal amphotericin B (L-AmB), and amphotericin B cholesteryl sulfate complex (ABCD) and exhibit a lower incidence of nephrotoxicity than that of AmB-d (12), although the safety profile of ABCD has limited its clinical use.
AmB must be administered parenterally because of its poor solubility and poor absorption when administered orally. It is widely distributed in various tissues and organs, including liver, spleen, bone marrow, kidney, and lung (13). Despite negligible concentration in cerebrospinal fluid (CSF), AmB formulations are effective in treating fungal infections of the central nervous system (CNS). In the past, intrathecal administration of AmB-d was used to treat meningeal infection (14,15); however, this practice is seldom used due to the difficulty of administration, poor patient tolerability, availability of alternative agents, and diminishing physician familiarity with this technique. The tissue distribution of the lipid formulations is similar to that of AmB-d; however, urinary excretion of lipid drugs is lower than that of AmB-d (16). All currently available formulations are highly protein bound (>95%, primarily to albumin) and have long half-lives.
AmB displays concentration-dependent fungicidal activity against many fungi with an optimal maximal concentration-to-mean inhibitory concentration (Cmax-to-MIC) ratio of 4–8:1 and a postantifungal effect of up to 12 hours (17). These findings suggest the peak serum level-to-MIC ratio is the best pharmacologic predictor of outcomes with polyene therapy. Drug levels are infrequently measured nor are necessary and are typically necessary only in the research setting (18).
AmB exhibits poor CSF levels (<5% of concurrent serum concentration); however, this agent remains the treatment of choice for cryptococcal meningitis (19). AmB formulations also have low vitreous penetration (0% to 38%) and intraocular injections may be required to achieve appropriate levels during therapy of deep ophthalmologic fungal infections including candidal endophthalmitis (20,21). The exact route of elimination of AmB is not known and despite the well-known nephrotoxicity, dosing need not be adjusted in patients with a decreased glomerular filtration rate (GFR).
AmB formulations are currently used primarily in the treatment of invasive cryptococcosis and moderate to severe infections with the endemic fungi (Histoplasma, Coccidioides, Blastomyces, etc.). Its use in the treatment of candidiasis and aspergillosis has been largely supplanted by other agents following landmark clinical trials (22,23). L-AmB is also commonly used in the treatment of febrile neutropenia refractory to broad-spectrum antibacterial agents (24). Histoplasmosis and cryptococcosis remain the only infections for which a lipid formulation of AmB (L-AmB) has demonstrated greater efficacy than the conventional form (25,26).
AmB formulations were previously the preferred first-line agent during the treatment of invasive aspergillosis (IA); however, a greater therapeutic response and survival have been demonstrated when voriconazole is administered in this setting—relegating AmB to second-line or salvage therapy during the treatment of IA (27). AmB does remain the agent of choice when Mucorales are encountered. In fact, a delay in the prescribing of an AmB formulation in patients infected with an agent of mucormycosis resulted in a twofold greater risk of death (28). Discriminating between invasive mucormycosis and aspergillosis is difficult, but the differences in the choice of antifungal agents and outcomes mandate an aggressive diagnostic strategy and prompt initiation of antifungal agents.
In attempts to avoid the potential nephrotoxicity of systemic administration and to deliver higher local concentrations, different formulations of AmB have been given via the inhalational route. AmB-d is often difficult to effectively administer in an aerosol form due to foaming caused by the solubilizing agent and the detergent-like effects are thought to possibly affect alveolar surfactant (28–31). Thus, lipid preparations are preferred for inhalation delivery. Aerosol delivery has been found effective in the prevention of pulmonary fungal infections in lung transplantation and in bone marrow transplant recipients, although data supporting its efficacy in other settings is limited (29).
Intravenous (IV) infusion of AmB-d is associated with reactions such as fever, chills, rigors, myalgias, bronchospasm, nausea and vomiting, tachycardia, tachypnea, and hypertension (32). These events are less likely to occur when one of the lipid formulations is used; however, ABCD has been associated with the development of dyspnea and hypoxia and L-AmB has been associated with back pain during infusion (20). AmB has been associated with acute kidney injury and nephrotoxicity in many studies and is a well-known potential complication of therapy occurring in up to 30% of patients. This toxicity is thought secondary to vascular smooth muscle dysfunction with resultant vasoconstriction and ischemia (33). For this reason, most advocate ensuring adequate volume status prior to administration. Lipid preparations of AmB have a lower incidence of renal toxicity, and studies have shown that when AmB-d is replaced by a lipid formulation after the development of creatinine elevation, renal function stabilizes or improves in a significant proportion of patients (34).
The avoidance of AmB-d and use of a lipid formulation has been met with skepticism by some due to the price difference in compounds. The reduction in hospital days when toxicity is avoided has proven the lipid formulations more cost-effective than AmB (34).
TRIAZOLES
The antifungal azoles include the imidazoles and the triazoles, which differ in terms of their chemical structure. Among the imidazoles (two nitrogens in the azole ring), only ketoconazole has systemic activity, and this agent has been replaced by more efficacious and less toxic alternatives and thus will not be discussed in this chapter. The triazoles (three nitrogens in the azole ring) all have systemic activity and include fluconazole, itraconazole, voriconazole, and posaconazole. An additional triazole, isavuconazole, is currently in phase III clinical trials.
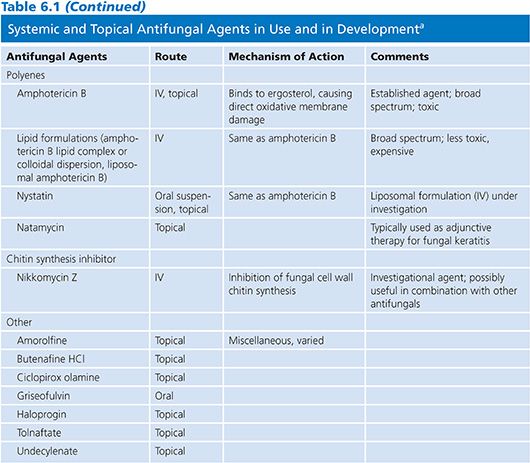
The triazoles also exert their effects within the fungal cell membrane. The inhibition of cytochrome P450 (CYP)–dependent 14-α-demethylase prevents the conversion of lanosterol to ergosterol. This mechanism results in the accumulation of toxic methylsterols and resultant inhibition of fungal cell growth and replication (Fig. 6.1). This class of agents has demonstrated both species- and strain-dependent fungistatic or fungicidal activity in vitro (35). Generally, these agents exhibit fungistatic activity against yeasts such as Candida and Cryptococcus; however, voriconazole appears to be fungicidal against Aspergillus spp. The area under the curve-to-MIC ratio (AUC/MIC) is the primary predictor of drug efficacy (36).
The indirect immunomodulatory effects are poorly understood due to the complex interaction of triazoles and phagocytic cells. Evidence suggests that ergosterol depletion increases fungal cell vulnerability to phagocytic oxidative damage (37) and voriconazole has been shown to induce the expression of TLR-2, nuclear factor-κB (NF-κB), and TNF-α (4).
Azoles differ in their affinity for the 14-α-demethylase enzyme and this difference is largely responsible for their varying antifungal potency and spectrum of activity. Cross-inhibition of several human CYP-dependent enzymes (3A4, 2C9, and 2C19) is responsible for the majority of the clinical side effects and drug interaction profiles that have been described with this class (38–41).
Itraconazole and posaconazole act primarily as inhibitors of 3A4 and 2C9 with little effect on 2C19. However, voriconazole acts as both an inhibitor and a substrate on all three isoenzymes, providing ample opportunity for drug–drug interactions due to this frequently shared metabolic pathway (41).
Comprehensive lists of triazole drug interactions can be found elsewhere (42). Briefly, caution should be used when these agents are concurrently administered with most HMG-CoA reductase inhibitors, benzodiazepines, phenytoin, carbamazepine, cyclosporine, tacrolimus, sirolimus, methylprednisolone, buspirone, alfentanil; the dihydropyridine calcium channel blockers verapamil and diltiazem; the sulfonylureas, rifampin, rifabutin, vincristine, busulfan, docetaxel, trimetrexate; and the protease inhibitors ritonavir, indinavir, and saquinavir (43–49).
The triazoles have also been associated with QTc prolongation (50) and coadministration with other agents known to have similar effects (cisapride, terfenadine, astemizole, mizolastine, dofetilide, quinidine, and pimozide, among others) should be avoided (51–53). The triazoles are additionally embryotoxic and teratogenic and are secreted into breast milk and thus, administration should be avoided during pregnancy or while lactating (50,54,55).
Fluconazole
Fluconazole remains one of the most frequently prescribed triazoles due to its excellent bioavailability, tolerability, and side effect profile. More than 80% of ingested drug is found in the circulation, protein binding is low (approximately 10%), and 60% to 70% is excreted unchanged in the urine (56). Oral absorption remains unchanged in patients receiving acid suppressive therapy (proton pump inhibitors or H2-blockers) (57).
Fluconazole is a water-soluble compound with excellent tissue penetration and is available in both oral and IV formulations. CSF levels are 50% to 70% of matched serum levels, and levels reported in saliva, sputum, and other sites are well within therapeutic ranges (58,59). The half-life is 27 to 34 hours in the presence of normal renal function, allowing once-daily dosing and it exhibits linear pharmacokinetics that are independent of doses and formulation. In patients with diminished creatinine clearance (CrCl), the normal dose should be reduced by 50% (38). Fluconazole exhibits concentration-independent fungistatic activity against Candida and Cryptococcus neoformans (60,61). The AUC/MIC ratio appears to be the most predictive pharmacodynamic parameter for fluconazole (62). Fluconazole serum levels are rarely necessary in clinical practice due to its predictable oral absorption and pharmacokinetic profile.
Fluconazole is active against most Candida spp with the exception of Candida krusei, which is intrinsically resistant to fluconazole (63). Candida glabrata is significantly less susceptible to fluconazole than Candida albicans, although wild-type isolates have MICs that extend to an MIC of more than 64 µg/mL. Previously, Candida glabrata strains were considered susceptible-dose dependent (S-DD at an MIC of 16 to 32 µg/mL), with roughly 10% exhibiting high-level resistance (MIC, more than 64 µg/mL) (21,64). A recent Clinical and Laboratory Standards Institute (CLSI) reclassification has resulted in the breakpoint for susceptible Candida glabrata being eliminated and strains considered S-DD at an MIC of less than or equal to 32 µg/mL. Fluconazole is active against most species of Candida, Cryptococcus neoformans, dermatophytes, Trichosporon spp, H. capsulatum, Coccidioides immitis, and P. brasiliensis (65,66). Only limited activity is seen against B. dermatitidis and resistance may develop when fluconazole is used to treat histoplasmosis (67). Fluconazole has no useful activity against molds including Aspergillus spp, Fusarium spp, or Mucorales (68).
Fluconazole has an important role in the treatment of candidiasis, cryptococcosis, and coccidioidomycosis (14,69,70). Fluconazole is used as primary therapy for candidemia and mucosal candidiasis, as well as prophylaxis in selected high-risk populations who are at lower risk for molds like Aspergillus (69). Patients with oropharyngeal candidiasis (OPC) are typically prescribed 100 mg/day for 7 to 14 days (21). Newer data suggests a one-time dose of 750 mg for the treatment of OPC with equivalent relapse rates to standard therapy (71). Patients with frequent relapse should remain on chronic suppressive fluconazole until immune reconstitution has been documented.
It is also used in maintenance therapy of cryptococcal meningitis in patients with AIDS (14,72). Following induction therapy with AmB and flucytosine, fluconazole is prescribed as an initial dose of 400 mg for 10 weeks followed by 200 mg weekly pending immune reconstitution (19). Although recent data has accrued regarding the use of high-dose fluconazole monotherapy during the induction course of cryptococcal meningitis, this practice should be used only in resource-limited settings and not when AmB is available (73).
Fluconazole is also the agent of choice in the treatment of disseminated coccidioidomycosis including meningitis. In these cases, high-dose fluconazole (up to 2 g daily) is often necessary (74). Similarly, other endemic mycoses respond favorable to fluconazole and this agent is considered second line during treatment of histoplasmosis and sporotrichosis (75,76).
Fluconazole has also been used for prophylaxis in those at high risk of invasive fungal infections. Initiation of 400 mg/day of fluconazole for the first 75 days following bone marrow transplantation (BMT) has been found effective in reducing cases of candidemia (77). Preemptive therapy in other settings, including within intensive care unit (ICU), remains controversial. The high incidence of invasive candidiasis within this setting (1% to 2% of all patients) makes prophylaxis an attractive option; however, the largest randomized, multicenter, blinded clinical trial comparing empiric fluconazole therapy to placebo in ICU patients with several risk factors for invasive candidiasis showed no clear benefit to fluconazole therapy (78).
Drug–drug interactions are less commonly observed with fluconazole than other triazole compounds, although caution remains necessary due to increases in the serum levels of phenytoin, glipizide, glyburide, warfarin, rifabutin, and cyclosporine. Fluconazole levels are reduced in the presence of rifampin (38).
Fluconazole is well tolerated by most patients, even if chronic therapy is necessary (79). Headache, alopecia, and anorexia are the side effects most common (10%), with transaminase elevation seen in fewer than 10%. Severe side effects such as exfoliative dermatitis and liver failure are very uncommon (38).
Itraconazole
Itraconazole is a lipophilic triazole currently available as both capsules and an oral solution suspended in hydroxypropyl-β-cyclodextrin (HP-β-CD) (39). The IV preparation of itraconazole is no longer commercially available.
The IV formulation provides optimal bioavailability with peak plasma levels obtained within 1 hour of administration. The HP-β-CD carrier is cleared renally and caution is recommended when administering the IV formulation to patients with impaired renal function (80). Itraconazole exhibits poor penetration into the CNS and a P-glycoprotein efflux mechanism has been found in mice to actively transport itraconazole out of the brain (81). The high protein binding (less than 1% available as free drug) and lipophilic nature of itraconazole also results in high concentrations in fatty tissues and purulent exudates (82).
Itraconazole exhibits concentration-independent fungistatic activity against Candida spp and Cryptococcus neoformans (83). In contrast, itraconazole has been shown to exert a time- and concentration-dependent fungicidal effect against Aspergillus spp (84).
The antifungal activity of itraconazole is broad and includes Candida spp, Cryptococcus spp, Aspergillus spp, dermatophytes, dematiaceous molds, Pseudallescheria boydii, Penicillium marneffei, B. dermatitidis, Coccidioides immitis, H. capsulatum, Paracoccidioides brasiliensis, and Sporothrix schenckii (Table 6.2). Itraconazole has activity against some, but not all, fluconazole-resistant strains of Candida krusei and Candida glabrata. Although rare, strains of Aspergillus fumigatus that are resistant to itraconazole have been reported and may be increasing in some regions (85,86). Mucorales, most strains of Fusarium, and Scedosporium prolificans are resistant to itraconazole (35).
Absorption of itraconazole capsules is erratic and requires an acidic gastric pH and administration with food. Itraconazole solution allows for greater oral bioavailability and the AUC and peak concentrations are both increased by 30% when itraconazole solution is taken in the fasting state (87,88). The cyclodextrin carrier has minimal absorption and no systemic side effects have been attributed to its use in the oral formulation (89). With once-daily dosing, steady state is reached in 7 to 14 days, although oral loading (200 mg three times daily for 3 days) allows for more rapid attainment of therapeutic serum levels (90).
Itraconazole is extensively metabolized by the liver and its major metabolite, hydroxyitraconazole, also possesses antifungal activity similar to that of the parent drug. Despite similar antifungal efficacy, hydroxyitraconazole is not measured during serum drug level determination by high-performance liquid chromatography (HPLC) and ultra-performance liquid chromatography with mass spectrometry (UPLC/MS) is instead required. The active metabolite is detected by bioassay (91).
The development of newer and more effective antifungal agents (i.e., voriconazole) has relegated itraconazole to second-line therapy during the treatment of IA. Itraconazole is thus licensed in the United States only for salvage therapy of IA and allergic bronchopulmonary aspergillosis (27).
Itraconazole remains the drug of choice for those with mild to moderate infection caused by histoplasmosis and is the mainstay of secondary prophylaxis in HIV patients with a history of histoplasmosis prior to immune reconstitution with antiretrovirals (75).
It is also useful for lymphocutaneous sporotrichosis and non–life-threatening, nonmeningeal forms of histoplasmosis, paracoccidioidomycosis, and blastomycosis (75,76,92–94), and nonmeningeal coccidioidomycosis and is the preferred agent for osseous manifestations of coccidioidomycosis (95). Additionally, itraconazole has been used for maintenance treatment of cryptococcal meningitis (14) and for some forms of phaeohyphomycosis (96). It has no activity against Fusarium spp, Mucorales, or S. prolificans (35).
The recommended dosage of oral itraconazole in adults is 400 mg/day (capsules) and 2.5 mg/kg twice daily (HP-β-CD solution) (27). However, steady-state levels can be more rapidly attained when administered as 200 mg three times daily for 3 days and then 200 mg twice daily for the duration of therapy. Considerable concern remains regarding adequate oral absorption and oral itraconazole is not recommended in seriously ill or patients with life-threatening disease. Dose adjustment is not indicated when the oral formulations of itraconazole is used in patients with renal insufficiency or those receiving hemodialysis/continuous ambulatory peritoneal dialysis (CAPD). The half-life of itraconazole is prolonged in patients with hepatic dysfunction and drug dose adjustment, liver function testing, and drug interactions should be carefully assessed (97).
Side effects due to itraconazole may occur including gastrointestinal intolerance, hypokalemia, edema, rash, and elevated transaminases (39). Severe hepatotoxicity is rare (98). In contrast to fluconazole, drug interactions are relatively common with itraconazole because of the inhibition of the oxidative metabolism of agents that are metabolized by hepatic cytochrome P450 enzymes (39,42).
Itraconazole is usually well tolerated, although adverse reactions have been observed in up to 39% of patients and dose-limited toxicity requiring discontinuation of therapy can occur. The most frequent side effects include nausea and vomiting (<10%), hypertriglyceridemia (9%), hypokalemia (6%), liver enzyme elevations (5%), skin rashes/pruritus (2%), headache and dizziness (<2%), and pedal edema (1%) (99). Gastrointestinal intolerance (46%) is exceedingly common with the oral HP-β-CD solution at doses greater than 400 mg per day, with vomiting as the most frequent complaint (100). The myocardial depressant effects of itraconazole are also well known and cases of congestive heart failure have been reported (101).
Posaconazole
Posaconazole is a lipophilic second-generation antifungal triazole with a similar molecular structure to that of itraconazole. However, posaconazole’s improved spectrum of activity also exhibits efficacy against Mucorales and has enhanced activity against Aspergillus spp compared to itraconazole (102).
Posaconazole is insoluble in water and an IV formulation is currently in phase III clinical trials while a solid oral formulation is also currently in development. It is currently administered as a cherry-flavored suspension using polysorbate 80 as the emulsifying agent (103). Optimal dosing of posaconazole is obtained when given as two to four divided doses administered with food or a liquid nutritional supplement (104,105). Although initial studies suggested gastric acidity did not affect posaconazole absorption subsequent work has shown H2-receptor antagonists and proton pump inhibitors may decrease posaconazole serum levels and coadministration should be avoided (54,103,106,107).
Posaconazole has demonstrated dose-dependent pharmacokinetics with saturable absorption above 800 mg per day. Oral loading is thus not possible and steady state is reached only after 7 to 10 days of therapy (108). This prolonged time required to reach steady-state levels also impacts enthusiasm for the use of posaconazole as primary therapy for invasive fungal infections. Posaconazole has a large volume of distribution, despite its high protein binding, and a half-life of approximately 24 hours.
Peak serum concentrations have shown considerable interpatient variability for reasons that remain unclear. Some have proposed genetic polymorphisms within P-glycoprotein play a role as posaconazole is both a substrate and inhibitor, but this remains unproven (109). Glucuronidation plays a minor role in posaconazole metabolism and single-nucleotide polymorphisms within UGT (uridine diphosphate-glucuronosyltransferase) have been proposed to account for these differences but confirmatory studies have not been performed (110). This unpredictable variation in serum posaconazole levels has heightened interest and the necessity of therapeutic drug monitoring (TDM).
Posaconazole is hepatically metabolized and, as discussed earlier, undergoes minimal glucuronidation. Renal clearance plays a minor role in the clearance of posaconazole, which is predominantly eliminated fecally.
Oral posaconazole has proven effective in the prevention of proven or probable IA in neutropenic patients with acute myelogenous leukemia (AML) and in hematopoietic stem cell transplant recipients with graft-versus-host disease (GVHD) (111,112). The efficacy and safety of posaconazole in the treatment of invasive fungal infections has also been assessed, and although this study predates the widespread use of echinocandins and voriconazole, the observed efficacy allows for its use during salvage therapy (113).
Currently, 200 mg three times daily is recommended for prophylaxis, and 800 mg divided in two or four doses is recommended in the salvage setting. Patients not tolerating food should be given a liquid nutritional supplement in attempts to increase drug absorption (109). Pediatric dosing schedules have yet to be definitively established yet retrospective data has demonstrated the safety and efficacy in this population (27,114). Dose adjustment by age, sex, race, and hepatic or renal insufficiency is not necessary given the minimal glucuronidation and renal clearance of posaconazole (115).
Posaconazole is typically well tolerated and infrequently requires discontinuation due to adverse drug events. The most frequent side effects of posaconazole therapy are gastrointestinal (14%), with transaminase elevation and hyperbilirubinemia occurring in 3% (112). However, in one trial, more serious adverse events were reported in patients treated with posaconazole than with fluconazole. Three cardiac events were reported among those possibly related to posaconazole treatment including decreased ejection fraction, QTc prolongation, and torsades de pointes (111). Nevertheless, for most patients, posaconazole is well tolerated and even long-term therapy (>6 months) is frequently without toxicity (116).
Posaconazole is not significantly metabolized through the cytochrome P450 system and serum levels are unlikely to be increased by concomitant administration of P450 inhibitors. Posaconazole is known to decrease the metabolism of other medications metabolized through these pathways and caution should be taken with coadministration.
Voriconazole
Voriconazole is a low-molecular-weight water-soluble extended-spectrum triazole with a chemical structure similar to fluconazole. Available in both oral and IV formulations, the latter is dependent on sulfobutyl ether β-cyclodextrin (SBECD) for solubility (117). The oral formulation of voriconazole is well absorbed with a bioavailability of more than 90% (118,119). Steady-state plasma levels range from 2 to 3 µg/mL following oral administration and 3 to 6 µg/mL after IV infusion. When 3 to 6 mg/kg of daily voriconazole is administered, steady-state levels are reached in 5 to 6 days. However, if oral or IV loading is given, steady state can be reached within 1 day (120).
Voriconazole is 58% protein-bound and has excellent penetration into the CNS as well as other tissues (121,122). It is metabolized in the liver via the cytochrome P450 enzyme family and less than 5% of voriconazole is excreted unchanged in urine (41,117).
Although voriconazole in children has demonstrated linear pharmacokinetics, in adults, nonlinear metabolism is observed, likely secondary to saturable metabolic enzymes required for drug clearance (120). Interpatient serum concentration differences have been attributed to polymorphisms within CYP2C19, the major metabolic pathway for voriconazole (117). Up to 20% of non–Indian Asians have low CYP2C19 activity and voriconazole serum levels are thus up to four times higher than those found in White or Black populations in which the “poor metabolizer” status is uncommon (123). The unpredictability of patient enzymatic activity has generated an increased interest in the routine use of voriconazole TDM.
For IV administration, 6 mg/kg twice daily on day 1, followed by 4 mg/kg IV twice daily for the duration of therapy is recommended. The oral dosages in adults are also weight based. For those weighing greater than 40 kg, 400 mg twice daily on day 1, followed by 200 mg twice daily until completion of therapy is suggested, whereas those weighing less than 40 kg should receive 200 mg twice daily for 1 day followed by 100 mg twice daily (123). Pediatric patients are known to hypermetabolize voriconazole and for this reason an IV dose of 7 mg/kg twice daily and oral dosing of 200 mg twice daily without loading is recommended (55). In patients with liver dysfunction, standard loading doses should be given, but the maintenance dose reduced by 50%. The safety of voriconazole use in severe liver disease remains uncertain. No dosage adjustment is required if oral drug is given to patients with renal insufficiency. However, the presence of a cyclodextrin vehicle within the IV formulation has caused concerns about vehicle accumulation in renal insufficiency or dialysis dependence and IV administration is best avoided in patients with a CrCl less than 50 mL (117).
Voriconazole exhibits concentration-independent fungistatic activity against Candida spp and Cryptococcus neoformans with no apparent post–antifungal effect (124). Recent in vivo studies of disseminated Candida albicans infection indicate that an AUC/MIC ratio of 20 to 25:1 predicts treatment success (125). In contrast, voriconazole appears to exert a dose-dependent fungicidal effect on Aspergillus based on both time-kill studies (126) and clearance of fungal burden in target organs of animal models of IA (127).
Voriconazole has a broad spectrum of activity that includes Candida spp, Cryptococcus neoformans, Trichosporon spp, Aspergillus spp, Fusarium spp, and other hyaline molds, dematiaceous fungi, and the endemic fungi. The anticandidal activity of voriconazole encompasses Candida krusei and most, but not all, strains of Candida albicans. Although activity of voriconazole in vitro against Candida glabrata can be demonstrated, the high MICs seen with wild-type Candida glabrata against voriconazole has resulted in no susceptible breakpoints for voriconazole against Candida glabrata in recent CLSI guidance (128,129). Voriconazole is also active against some fungi that are resistant to AmB, including A. terreus and P. boydii (130,131). Voriconazole exhibits a broad spectrum of activity against molds with the exception of agents of mucormycosis (117), and in fact, Mucorales have been proven more virulent if exposed to voriconazole (132).
Voriconazole is approved for the primary treatment of IA (22,133) and for treatment of infections due to P. boydii (Scedosporium apiospermum) and Fusarium spp in patients intolerant of, or with infections refractory to, other antifungal agents (134). Importantly, voriconazole was shown to be more effective than AmB (53% vs. 32% complete or partial response at week 12 of treatment and 71% vs. 58% survival, respectively) for primary treatment of IA (23). Voriconazole has also been shown to have good efficacy in the treatment of various forms of candidiasis (135,136). It has not been approved for empirical treatment of febrile, neutropenic patients despite documented efficacy in the treatment of IA and in the prevention of breakthrough fungal infections in this same patient population (137).
Typically well tolerated, the side effect profile of voriconazole is similar to other triazoles with a few notable exceptions. The majority of those experiencing a reported adverse reaction to voriconazole describe abnormal vision (up to 23%) that is transient, infusion related, and without long-term sequelae (138). This unique effect typically occurs 30 minutes after infusion and abates 30 minutes after onset.
Other well-known effects of voriconazole therapy include skin rash and transaminase elevation in 10% to 20% of recipients (133). Baseline evaluation of hepatic function has been recommended before and during treatment, and rare cases of hepatic failure during voriconazole use have been reported (139). Elevated voriconazole serum levels have been attributed to the majority of side effects encountered in clinical practice, and higher levels (>5.5 mg/L), although associated with favorable outcomes, have also been suggested responsible for the uncommon potential side effects of encephalopathy or hallucinations (140–142).
Therapeutic Drug Monitoring for Azole Antifungals
Commercial assays are available for monitoring the serum concentrations of all currently available triazoles; however, at this time, existing guidelines recommend only itraconazole TDM (75,143,144). A strong argument can also be made for routine TDM of both posaconazole and voriconazole as well.
Itraconazole levels should be drawn after steady state is reached to ensure therapeutic levels (>1 µg/mL). Fluconazole levels are infrequently monitored due to the excellent bioavailability of this agent. However, clinical circumstances may dictate drug monitoring when therapeutic levels are uncertain (i.e., concurrent use of rifampin, rifampicin, etc.).
The extended-spectrum triazoles, posaconazole and voriconazole, have received increased attention due to their erratic absorption (posaconazole), or concerns for toxicity and the interpatient variability of serum levels (voriconazole). No guidelines exist for posaconazole TDM; however, past evidence supports a relationship between posaconazole serum drug level and efficacy (145). TDM should also be considered when drug interactions are of concern, such as the aforementioned potential for acid-suppressive agents to reduce absorption, although goal levels remain to be determined. Most experts suggest target trough concentrations greater than or equal to 0.5 µg/mL when given for antifungal prophylaxis. The interpatient variability of voriconazole also warrants consideration of TDM during use. Low concentrations (<1 mg/L) are more common in those receiving oral therapy and failure rates have been associated with low serum drug levels (133). Conversely, levels greater than 5.5 mg/L have been associated with encephalopathy without an improvement in efficacy (140). However, the frequency with which to monitor these newer triazoles remains to be determined.
ECHINOCANDINS
Echinocandins (caspofungin, micafungin, anidulafungin) are semisynthetic lipopeptides that inhibit the synthesis of β-1,3 glucan, an important constituent of the fungal cell wall (see Fig. 6.1), by inhibiting the activity of glucan synthase. The glucans are important in maintaining the osmotic integrity of the fungal cell and play a key role in cell division and cell growth (1,146). Disruption of β-1,3 glucan synthesis thus impairs cell wall integrity and leads to osmotic lysis (147).
The echinocandins are currently available only in IV formulations. The echinocandins exhibit dose-dependent linear pharmacokinetics and are highly (more than 95%) protein bound (148–150). They are broadly distributed to all major organs, including the brain (151), although concentrations in CSF are low (152). Metabolism occurs in the liver by a cytochrome P450 independent mechanism and the inactive metabolites are excreted in the feces and urine; less than 2% of a dose is excreted in the urine in active form (153,154).
Both in vitro and in vivo studies in Candida spp have shown a concentration-dependent fungicidal effect that is optimized at a peak-to-MIC ratio of approximately 8:1 (18,155) and a postantifungal effect of up to 12 hours (156). In contrast, activity against Aspergillus spp is neither classically fungistatic nor fungicidal, with activity localized to the growing hyphal tips and branch points with resultant inhibition of growth and angioinvasion but only a modest effect on the fungal burden in tissues (157–159).
TDM of echinocandins is almost never required and not recommended. Administered as standard doses, this class of antifungals has recently received attention for dosing strategies that may be needed in those with morbid obesity, although definitive recommendations have not been made (160,161).
Multiple in vitro studies have confirmed a paradoxical effect of the echinocandins. In this circumstance above a certain concentration of drug decreased antifungal activity is observed. The exact mechanism responsible for this phenomenon has not been fully elucidated, although studies have shown the involvement of the protein kinase C cell wall integrity pathway and an increase in cell wall chitin content as potential mechanisms responsible for this phenomenon (162). The clinical significance of these in vitro findings remains uncertain (163) and higher echinocandin doses have been evaluated in prospective trials and outcomes are no different from those at standard dosing (164).
Echinocandins have poor oral absorption and current agents are available only in the IV formulation. Echinocandins are highly protein bound (anidulafungin 98%, caspofungin 96%, and micafungin 99.8%) and have a half-life of 40 hours, 10.6 hours, and 11 to 17 hours, respectively (80). Their vitreal and CSF penetration is negligible and this point is of clinical significance during the treatment of candidemia if endophthalmitis is also observed (165).
Caspofungin was the first available agent of this class and is metabolized by both hepatic hydrolysis and N-acetylation. Inactive metabolites are subsequently eliminated in the urine. Severe hepatic dysfunction thus mandates caspofungin dose reduction (20). Caspofungin has several drug interactions with agents metabolized through the cytochrome P450 system and serum levels are reduced in the presence of rifampin and may increase levels of sirolimus, nifedipine, and cyclosporine (20). Micafungin is metabolized by nonoxidative metabolism within the liver and anidulafungin undergoes nonenzymatic degradation within the kidney. Both agents are eliminated in feces. These agents therefore do not require dosage adjustment with hepatic impairment (20).
Their clinical use is primarily limited to Candida spp and Aspergillus spp and they lack activity against Mucorales, Cryptococcus spp, and other clinically important molds (Table 6.1). Although activity is observed against all Candida spp, the mean inhibitory concentrations (MICs) are elevated (>1 µg/mL) when Candida parapsilosis and Candida guilliermondii are encountered. Susceptibility differences between the different agents in this class are minimal (166). Recent changes in CLSI breakpoints recognize that although clinical activity of the echinocandins is good even with higher MICs, mutations in the target FKS1/2 gene are likely present with MICs over 0.25 µg/mL for most species with higher breakpoints for C. parapsilosis and C. guilliermondii. Echinocandins also have immunomodulatory effects. By exposing β-glucan by the disruption of fungal cell wall mannoproteins, additional antigens are exposed for antibody deposition and fungal recognition by the host immune system, which may add activity against organisms such as molds like Mucorales and Fusarium for which they have minimal intrinsic activity (167).
In addition to activity against Candida and Aspergillus spp, the echinocandins have moderate activity against dematiaceous fungi (168,169) and low activity against the endemic dimorphic pathogens (169,170). They are inactive against Fusarium and other hyalohyphomycetes, Cryptococcus neoformans, Trichosporum spp, and Mucorales (150,171). Primary resistance to echinocandins appears to be uncommon in Candida albicans and Aspergillus spp and notably, echinocandins are active against fluconazole-resistant strains of Candida spp (32,172,173).
The increased incidence of triazole-resistant Candida spp and the fungicidal activity of the echinocandins (caspofungin, micafungin, and anidulafungin) have prompted some authorities to recommend these agents as first-line therapy for invasive candidiasis and recent meta-analysis has shown improved outcomes in those who receive echinocandins as first-line therapy (174). Additionally, their proven efficacy, infrequency of side effects, and favorable drug interaction profiles make them attractive options over other available antifungals (32,164,175,176).
Comparative trials have found the echinocandins equally efficacious and better tolerated than AmB in the treatment of candidemia (32). In one such trial, caspofungin (70 mg loading dose followed by 50 mg daily) was compared to AmB-d (0.6 to 1 mg/kg) in the treatment of invasive candidiasis. Although C. albicans was more common in the AmB arm, modified intention to treat analysis revealed similar survival in each group, with a trend toward increased survival and a statistically significant decrease in drug side effects in those receiving caspofungin (32).
Similarly, micafungin (100 mg IV daily) has been compared to L-AmB 3 mg/kg IV daily in an international, double-blind trial. In this study assigning patients to 14 days of IV treatment, successful treatment was equivalent in each group. However, there were fewer treatment-related adverse events with micafungin than there were with liposomal amphotericin (175).
Only one comparative trial of different echinocandins has been performed in invasive candidiasis. In this trial, patients were enrolled to one of three treatment groups: micafungin 100 mg/day IV daily, micafungin 150 mg IV daily, or caspofungin 70 mg IV loading dose followed by 50 mg IV daily. No differences were found between treatment groups including microbiologic failure or all-cause mortality (164). Although this trial found that higher doses of an echinocandin may not equate to a greater therapeutic response, no increase in toxicity was seen with higher doses nor was a paradoxical effect observed.
Anidulafungin has been compared with fluconazole for the treatment of invasive candidiasis and treatment was successful in 75.6% of patients treated with anidulafungin, as compared with 60.2% of those treated with fluconazole. Survival was improved with anidulafungin but was not statistically significant compared to the fluconazole arm (176).
Collectively, these trials have shown a high level of efficacy and low toxicity for the echinocandins. This class has since become “first-line” therapy in the treatment of candidiasis in most medical centers pending species identification and documented clinical improvement in the patient. After clinical improvement is obtained or the absence of fluconazole resistance documented, therapy is often changed to a triazole such as fluconazole (69). As noted earlier, CNS and intraocular infections should not be treated with echinocandin monotherapy due to their poor penetration into these sites.
Although clinical trials have been primarily limited to patients with candidemia, observational data has shown efficacy in candidal osteomyelitis, peritoneal infections, and abdominal abscesses (177). Additional retrospective data has also shown echinocandins may play a role in the treatment of infective endocarditis caused by Candida spp (178).
The echinocandins have also been found efficacious in the treatment of IA, although they are fungistatic against this genus. The known toxicity of AmB and its different formulations and the potential for voriconazole-induced drug–drug interactions or toxicity has increased interest in the echinocandins for use during treatment of IA (179).
Caspofungin as a potential first-line agent for IA has been evaluated in limited settings but data is not sufficient to recommend caspofungin or any of the echinocandins for first-line therapy in the treatment of IA. Thus, these agents are currently recommended in the treatment of IA only in those patients refractory to or intolerant of other agents (27,179). In vitro studies and limited clinical data have also shown the potential role for combination therapy (an echinocandin plus AmB or an azole) and a prospective trial of anidulafungin and voriconazole has been completed, with results suggesting benefit in subpopulations of patients but not in the entire population studied, although at the present time, full publication of the trial data has not occurred.
The side-effect profile of the echinocandins is very favorable and these agents are typically well tolerated. The most frequently reported adverse effects include increased liver transaminases, gastrointestinal upset, and headache (32,153). An infusion-related reaction has been described if rapid administration is given with tachycardia, hypotension, and/or thrombophlebitis. Drug interactions are rare; however, concomitant administration of caspofungin and cyclosporine is not recommended, yet reports have shown it may be safe to do so in selected cases (180,181). Cyclosporine has been noted to increase the AUC of caspofungin by approximately 35%, but these alterations are not sufficient to suggest dosage modifications (150). The authors do not typically make dose adjustments of the echinocandins in any of these situations.
ANTIMETABOLITES
Flucytosine
Flucytosine (5-FC) is a water-soluble, synthetic, fluorinated pyrimidine analogue that exerts antifungal activity by interfering with the synthesis of DNA, RNA, and proteins in the fungal cell. In vivo, 5-FC is taken up into the cell by a fungus-specific cytosine permease and deaminated in the cytoplasm to 5-fluorouracil (5-FU) by cytosine deaminase. 5-FU is further converted to 5-fluorodeoxyuridylic acid, which interferes with DNA synthesis. Mammalian cells lack cytosine deaminase, allowing for a selective inhibition of fungal organisms (see Fig. 6.1) (182). This agent may be either fungistatic or fungicidal, depending on both fungal species and strain.
5-FC is available in an oral formulation with excellent bioavailability. Unfortunately, the IV formulation is no longer widely available. High concentrations of 5-FC may be achieved in serum, CSF, and other body fluids (183). In vitro pharmacodynamic studies have determined that 5-FC exhibits concentration-independent fungistatic activity against Candida spp that is optimized at concentrations four times the MIC (184). Furthermore, 5-FC exerts a significant post–antifungal effect of 2.5 to 4 hours against Candida spp (184). These findings have been confirmed in vivo by Andes and van Ogtrop (185), who showed that time above the MIC was the pharmacodynamic parameter that correlated best with outcome in 5-FC treatment of murine candidiasis. Maximum efficacy was seen when 5-FC blood levels exceeded the MIC for only 20% to 25% of the dosing interval (185). The later observation may be accounted for by the post–antifungal effect of 5-FC.
5-FC has excellent oral bioavailability with over 80% to 90% absorption. Peak serum levels occur 1 to 2 hours after ingestion (30 to 45 µg/mL) of a single dose. The volume of distribution (Vd) of 5-FC is 0.6 to 0.9, yet bone, peritoneal, and synovial fluid 5-FC levels have been demonstrated and urinary levels are several folds higher than concurrent serum levels. Greater than 95% of 5-FC is eliminated unchanged in the urine. 5-FC is typically administered by mouth at 100 mg/kg/day divided in four doses (80) and that that dose is usually well tolerated without the need to adjust the dose based on drug levels.
Activity has been observed against most fungal pathogens including Candida, Cryptococcus, Cladosporium, Phialophora, and Saccharomyces spp. However, Aspergillus spp, Mucorales, dermatophytes, and the endemic mycoses are all resistant to 5-FC (1). Additionally, resistance commonly develops when 5-FC is used as monotherapy even in susceptible organisms and it should not be used as such except during the treatment of chromoblastomycoses or during the treatment of localized candidal infections when alternative agents are unavailable or contraindicated.
5-FC is primarily used only in the treatment of cryptococcus (combined with AmB) and chromoblastomycosis. Despite concerns for additive toxicity, the synergistic effects of dual therapy in Cryptococcus allow for more rapid CSF clearance (186).
Peak plasma levels of 40 µg/mL easily exceed the MICs of most Candida spp and Cryptococcus neoformans (187,188) and provide optimal antifungal activity while minimizing hematologic adverse effects (189). The marrow suppressive effects are more common if blood levels exceed 100 to 125 µg/mL (190). In the presence of prolonged therapy (>7 days) or with alterations in renal function, serum drug monitoring can be considered but at the lower doses typically recommended for use, drug level monitoring is not frequently necessary. Other less common side effects such as abdominal pain or diarrhea are frequently indirect markers of elevated 5-FC levels and therapy is typically stopped in these circumstances. Rash and hepatic transaminase elevation have also been reported with liver abnormalities frequently observed (183). 5-FC is teratogenic and should not be administered during pregnancy.
Allylamines
The allylamine class of antifungal agents is composed of terbinafine and naftifine. Only terbinafine has systemic activity. Both terbinafine and naftifine inhibit the enzyme squalene epoxidase, which results in the accumulation of squalene and blocks the synthesis of ergosterol (191). The accumulation of high concentrations of squalene results in increased membrane permeability and, ultimately, cell death.
Terbinafine
Terbinafine is a lipophilic antifungal agent that is available in oral and topical formulations (192). Terbinafine has a broad spectrum of activity that includes dermatophytes, Candida spp, Malassezia furfur, Aspergillus spp, Cryptococcus neoformans, Trichosporon spp, S. schenckii, and P. marneffei (193–196).
Terbinafine is well absorbed orally and is concentrated in fatty tissues, skin, hair, and nails but low serum levels of drug are found (197). Low toxicity and good efficacy in the treatment of virtually all dermatomycoses, including onychomycosis, is seen with terbinafine (198,199). The combination of terbinafine and fluconazole has shown efficacy in the treatment of fluconazole-resistant Candida spp yet with the recent availability of alternative antifungals, other agents are typically chosen (200). Terbinafine also has shown clinical efficacy in cases of sporotrichosis, aspergillosis, and chromoblastomycosis (201).
It is an inhibitor of CYP2D6, but this generally results in less important drug–drug interactions than inhibitors of CYP3A4. The most commonly reported side effects include gastrointestinal upset and cutaneous reactions (202). Reversible agranulocytosis has also been reported but is rare (203,204). Hepatic dysfunction and even fulminant hepatic failure are uncommon but have been reported during terbinafine administration and for this reason, terbinafine is not recommended in those with underlying liver disease (202).
Griseofulvin
Griseofulvin is a long-standing oral agent used for the treatment of dermatomycoses. Its mechanism of action is thought to involve interaction with microtubules within the fungal cell and inhibition of mitosis (205,206).
Griseofulvin is active against most dermatophytes; however, alternative agents, such as itraconazole and terbinafine, appear to be more potent and exhibit greater efficacy in prospective clinical trials (207–210).
Griseofulvin is best absorbed orally when administered in an ultramicrocrystalline form with an accompanying meal. It is deposited primarily in keratin precursor cells. Administration of griseofulvin is associated with a number of mild side effects including nausea, diarrhea, headache, cutaneous disruptions, hepatotoxicity, and neurologic complaints (211).
ANTIFUNGAL AGENTS FOR TOPICAL USE
There are numerous topical antifungal preparations available for the treatment of superficial cutaneous and mucosal fungal infections. The available preparations encompass a wide variety of antifungal classes including polyenes (AmB, nystatin, and natamycin), allylamines (naftifine and terbinafine), and numerous imidazoles and other miscellaneous agents. Preparations for use in cutaneous disease and onychomycosis include creams, lotions, ointments, powders, and sprays, whereas suspensions, tablets, troches, and suppositories are used for treatment of various forms of mucosal candidiasis (69,212,213).
The selection of topical versus systemic therapy for cutaneous or mucosal fungal infections depends on the status of the host and the type and extent of the infection. The refractory nature of infections such as onychomycosis or tinea capitis usually mandates long-term systemic therapy, whereas most cutaneous dermatophytic infections and oral or vaginal thrush respond favorably to topical therapy. Systemic agents are generally recommended for chronic, recurrent oral or vaginal candidiasis and for candidal esophagitis (69,213).
ANTIFUNGAL AGENTS UNDER DEVELOPMENT
Currently, several different antifungal agents are under active development, including those with established modes of action as well as novel new classes of antifungal agents (Table 6.1). These include a novel triazole agent (isavuconazole), IV and tablet posaconazole formulations, oral echinocandins, a chitin synthase inhibitor (nikkomycin Z), and agents with new mechanisms of action such as blocking critical or unique fungal cell pathways, such as elongation factor, fungal gene methylation, chitin synthesis, and others. The mechanisms of action and spectra of activity of the novel triazoles and the echinocandins are essentially the same as currently available agents. In each case, the newer agents in the respective classes offer the potential of fewer toxicities and drug interactions, more favorable pharmacokinetic and pharmacodynamic properties, and possible improved activity against selected refractory pathogens. Nikkomycin Z, which inhibits chitin synthesis in the fungal cell wall, provides another novel mode of action that may act in concert with other inhibitors of cell wall or cell membrane synthesis. The introduction of several of these new agents such as isavuconazole, which is in active clinical development and the new formulations of posaconazole, should augment our available antifungal resources. The development of agents with novel modes of action is both necessary and promising for future antifungal therapy.
MECHANISMS OF RESISTANCE TO ANTIFUNGAL AGENTS
Resistance to antimicrobial agents is an issue of concern worldwide with important implications for morbidity, mortality, and the costs of health care. Although much attention has been focused on antibacterial resistance (214), innate or acquired resistance to antifungal agents is now recognized among several pathogenic fungi (215,219). This recognition has spurred the development of new antifungal agents as well as intensive efforts to define the molecular mechanisms of resistance to various agents (Table 6.3).
The extensive utilization of fluconazole has been coupled with increasing reports of azole resistance (216,217) and some of the most elegant investigations of antifungal resistance mechanisms have involved the azole class of antifungals and Candida spp (218). In this section, we will review what is known of the molecular and cellular mechanisms of resistance to antifungal agents as well as the clinical factors that may result in resistance to antifungal therapy.
Molecular and Cellular Mechanisms of Resistance to Antifungal Agents
Most fungal infections are caused by Candida spp and most of our understanding of the mechanisms of resistance comes from studies of Candida albicans and other species of Candida (218–220). Although Aspergillus spp and Cryptococcus neoformans constitute a significant proportion of opportunistic mycoses, fewer studies have been performed on these organisms and little information on antifungal resistance mechanisms is available for other opportunistic fungal pathogens (68).
Although there are many parallels that exist between antibacterial resistance mechanisms and antifungal resistance mechanisms (191), there are no data to suggest that destruction or modification of antifungal agents is an important component of antifungal resistance. Likewise, it does not appear that fungi can employ the genetic exchange mechanisms that allow rapid transmission of antimicrobial resistance in bacteria. On the other hand, it is apparent that multidrug efflux pumps, target alterations, and reduced access to targets are important mechanisms of resistance to antifungal agents, just as they are important in antibacterial resistance (220,221). In contrast to the rapid emergence of high-level antimicrobial resistance that occurs among bacteria, antifungal resistance usually develops slowly and involves the emergence of intrinsically resistant species or a gradual, stepwise alteration of cellular structures or functions that results in resistance to an agent to which there has been prior exposure (221,222).
Polyenes
Resistance to AmB remains uncommon despite extensive utilization over more than 40 years. Among the Candida spp, decreased susceptibility to AmB has been reported in C. lusitaniae, C. glabrata, C. krusei, and C. guilliermondii (219,223–226). Although primary resistance to AmB is seen chiefly among C. lusitaniae, C. krusei, and C. guilliermondii (68,227,228). Most reports of AmB resistance in Candida spp appear to be secondary to AmB exposure during treatment (219,223).
Most isolates of Aspergillus spp appear susceptible to AmB; however, A. terreus seems to be resistant to AmB both in vitro and in vivo (229,230). Similarly, some of the “cryptic” strains of Aspergillus such as Aspergillus lentulus and others such as Aspergillus calidoustus may show decreased amphotericin susceptibility. Primary resistance to AmB among C. neoformans has not been reported while secondary resistance appears rare (187,231,232).
Our understanding of the mechanism of resistance to AmB stems largely from studies of mutants of Candida spp and Cryptococcus neoformans derived from sequential passage in varying concentrations of AmB (233) and from characterization of serial isolates from patients failing AmB therapy (218,223,231,234–238). The mechanism of AmB resistance appears to be from a qualitative or quantitative alteration in the sterol content of cells (239,240), defense mechanisms against oxidative damage (241), defects in ergosterol biosynthetic genes (242), and alterations in the sterol-to-phospholipid ratio (239).
Ergosterol is the primary sterol target for AmB in the fungal cell membrane and resistant yeast containing an altered sterol content bind lesser amounts of AmB than do susceptible cells. Accordingly, mutants of Candida spp and Cryptococcus neoformans resistant to AmB have been shown to have a reduced total ergosterol content (231,235), replacement of polyene-binding sterols (ergosterol) by ones that bind polyenes less well (fecosterol) (231,243), or masking of ergosterol in the cell membrane so that binding with polyenes is hindered by steric or thermodynamic factors (191,244). One or more of these factors may account for decreased susceptibility to AmB. This resistance is often specific for AmB, but cross-resistance to azoles has also been reported and (5,6)-desaturase (Erg3p) mutants identified as causal in isolates with resistance to both classes (223,245). The molecular mechanisms of AmB resistance in Candida and Cryptococcus neoformans have not been determined; however, sterol analyses of resistant isolates suggest that they are defective in erg2 or erg3, genes encoding for the C-8 sterol isomerase and C-5 sterol desaturase enzymes, respectively (218).
Azoles
The excellent safety profile of fluconazole has led to extensive utilization of this agent worldwide. Concomitant with this utilization reports of emerging resistance to fluconazole and other azoles have appeared (246). Despite these reports, primary resistance to fluconazole is unusual among most species of Candida causing bloodstream infections (216). Among the five most common species of Candida isolated from blood (C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, and C. krusei), the overall frequency of high-level resistance (previously reported as an MIC >64 µg/mL) to fluconazole is less than 3%. Resistance is uncommon among bloodstream infections of C. albicans (0% to 7%) (216,247), C. parapsilosis (0% to 4%), and C. tropicalis (0% to 6%), whereas approximately 12% or more of C. glabrata isolates exhibit primary resistance to this agent (247,248). C. krusei is considered intrinsically resistant to fluconazole and MICs are usually more than 32 µg/mL for this species (36,249). Notably, the frequency of resistance to fluconazole among bloodstream infection isolates of Candida spp has not increased substantially after more than two decades of utilization worldwide (216,247,250).
The new triazoles (posaconazole and voriconazole) exhibit more potent activity against Candida spp than that of fluconazole, including activity against C. krusei and some fluconazole-resistant strains of Candida (251–255). Isolates of C.glabrata for which fluconazole MICs are less than 16 µg/mL typically have MICs less than 1 µg/mL to voriconazole and posaconazole (129,256,257); however, those isolates with higher fluconazole MICs tend to be less susceptible to the new triazoles with MICs more than 2 µg/mL (129,256). Recent breakpoints for voriconazole do not include a susceptible category for voriconazole for C. glabrata and posaconazole breakpoints have not yet been established. In general, among Candida isolates, there is a strong positive correlation between fluconazole MICs and those of voriconazole and posaconazole, suggesting significance cross-resistance, especially for some species such as C. glabrata (129,254).
Similar to Candida spp, primary resistance to fluconazole is uncommon among isolates of Cryptococcus neoformans (187,258,259), although secondary resistance has been described among individuals with AIDS and relapsing cryptococcal meningitis (260). Despite these reports, the susceptibility profile of this organism to fluconazole has remained unchanged over the past 30 years (187,260).
Although Aspergillus spp are intrinsically resistant to fluconazole, most isolates appear susceptible to itraconazole and the new triazoles (261). MICs greater than 1 µg/mL for these azoles are unusual among clinical isolates of Aspergillus, although the incidence may be increasing in certain regions due to environmental use of triazole fungicides (262,263). In contrast to Candida, cross-resistance between itraconazole and the new triazoles is not complete: cross-resistance between itraconazole and posaconazole, but not voriconazole, has been observed in some strains (264).
The mechanism of azole resistance in Candida has been extensively evaluated for fluconazole and C. albicans (218,221). Resistance can result from a modification in the quality or quantity of the target enzyme, reduced access of the drug to the target, or some combination of these mechanisms. In the first instance, point mutations in the gene (ERG11) encoding for the target enzyme, 14-α-demethylase, leads to an altered target with decreased affinity for azoles. Overexpression of ERG11 results in the production of high concentrations of the target enzyme, creating the need for higher intracellular azole concentrations to inhibit increased available substrate. Loss of allelic variation in the ERG11 promoter may also result in a resistant strain that is homozygous for the mutated gene (265).
The second major mechanism involves active efflux of azole antifungal agents out of the cell through the action of two types of multidrug efflux transporters: the major facilitators (encoded by MDR genes) and those of the ATP-binding cassette superfamily (encoded by CDR genes) (219). Upregulation of the MDR1 gene leads to fluconazole resistance, whereas upregulation of CDR genes leads to resistance to multiple azoles (221,266–269). Evidence that these mechanisms may act individually, sequentially, and in concert has been derived by studying serial isolates of C. albicans from AIDS patients with OPC (270–272).
It appears that the mechanisms of resistance to azoles in C. glabrata involve upregulation of CDR1 genes, resulting in resistance to multiple azoles (266). Azole resistance in C. krusei appears to be mediated by reduced susceptibility of the target enzyme to inhibition by fluconazole and itraconazole (63). This does not seem to be the case with the new triazoles, given their potent aforementioned activity against C. krusei.
In Aspergillus, the azole target cyp51A is an established “hot spot” for mutations that confer phenotypic triazole resistance (273). A substitution of leucine 98 for histidine in the cyp51A gene, together with two copies of a 34-bp sequence in tandem in the gene promoter (TR/L98H), was found to be the dominant resistance mechanism. Recently, a mutation in the transcription factor complex subunit HapE has also been reported (274) and with the advent of whole genome sequencing, other mechanisms are likely to be seen in the near future.
The C. neoformans isolates that have developed secondary resistance to fluconazole have been shown to have an altered target enzyme or overexpression of MDR efflux pumps (275,276). The latter mechanism, termed heteroresistance, is intrinsic in all cryptococcal isolates and is easily induced in vitro (277).
Echinocandins
Caspofungin, anidulafungin, and micafungin all exhibit potent fungicidal activity against most species of Candida, including azole-resistant strains (18,278,279). Isolates for which echinocandin MICs exceed 1 µg/mL rarely occur outside of C. parapsilosis and C. guilliermondii; however, recent evidence has suggested the incidence of echinocandins resistance in C. glabrata may be increasing (216). Efforts to produce laboratory mutants of Candida spp with reduced susceptibility to caspofungin have demonstrated that the frequency of these mutants is extremely low (1 in 108 cells), suggesting a low potential for the emergence of resistance in the clinical setting (280,281); however, the development of resistance despite continuous receipt of an echinocandins has been reported (282). Likewise, isolates of Aspergillus spp from clinical sources with reduced susceptibility to echinocandins have been observed, although this appears an uncommon occurrence (171,283).
Studies of laboratory-derived mutants of C. albicans with reduced in vitro and in vivo susceptibility to the echinocandins have documented point mutations in the FKS1 and FKS2 genes encoding for the glucan-synthesis enzyme complex (282,284,285). These mutant strains demonstrate an increased 50% inhibitory concentration (IC50) for inhibition of the glucan synthesis enzyme complex and reduced susceptibility to all echinocandins in vitro and in vivo in animal models (280,281,286). These strains remain susceptible to polyenes and azole antifungal agents, although multidrug resistance to both echinocandins and azoles has been reported with C. glabrata. FKS genes are similarly essential in Aspergillus spp (287), and increased expression of the FKS gene has been observed in a resistant isolate (283), whereas other strains have been found to have mutations in the ECM33 gene (AfuEcm33), encoding cell wall proteins important for fungal cell wall organization (288,289). Although there are limited data currently, these observations suggest that while mutations in the hot spot regions of the FKS gene is the predominant resistance mechanism in Candida, it is possible that mechanisms outside the target gene may be more important in Aspergillus.
Flucytosine
Despite reports in the older literature of a higher frequency of primary resistance to 5-FC among Candida spp and Cryptococcus neoformans (290,291), more recent studies using validated, standardized test methods indicate that primary resistance is actually uncommon among bloodstream infection isolates of both Candida spp and Cryptococcus (187,188). Secondary resistance to 5-FC, on the other hand, is well documented to occur among both Candida spp and Cryptococcus neoformans during monotherapy with this agent (292).
Resistance to 5-FC may develop from decreased uptake (loss of permease activity) or by loss of enzymatic activity required for the conversion of 5-FC to 5-FU (cytosine deaminase) and 5-fluorouridylic acid (FUMP pyrophosphorylase) (292,293). Of these possible mechanisms, the most important appear to be the loss of cytosine deaminase activity or the loss of UMP pyrophosphorylase activity. Uracil phosphoribosyltransferase, another enzyme in the primidine salvage pathway, is also important in the formation of FUMP and loss of its activity is sufficient to confer resistance to 5-FC (293).
Allylamines
Allylamine resistance has been infrequently reported despite clinical failures in close to 25% of patients treated with these agents (191). Laboratory-created resistant isolates have been found to carry extra copies of the ERG1 gene (294) or single base pair exchanges in the ERG1 gene coding for squalene epoxidase, the target of terbinafine (295). Subsequently, clinical isolates with amino acid substitutions within the ERG1 gene have also been reported (296,297). Sanglard et al. (267) have reported that the CDR1 multidrug efflux pump can use terbinafine as a substrate, thus the possibility of efflux-mediated resistance to allylamines exists.
Clinical Factors Contributing to Antifungal Resistance
Fungal infections may fail to respond to appropriate antifungal therapy, thereby demonstrating “clinical resistance,” despite the fact that the drug employed is active against the infecting organism. The interaction of the host, the drug, and the fungus is complex and clinical outcomes are influenced by a number of interactions (298).
During treatment of invasive fungal infections, the immune status of the host is the most important factor in determining outcomes. The presence of neutrophils, utilization of immunomodulating drugs, concomitant infections (e.g., HIV), surgical procedures, age, and nutritional status all may be more important than the ability, or lack thereof, of antifungal agents to inhibit or kill the infecting organism. Likewise, the site and severity of infection plays a critical role in the pharmacokinetic/pharmacodynamic interactions during antifungal therapy. The presence of a foreign body, such as catheter, prosthetic valve, or vascular graft material, may allow an otherwise susceptible organism to cause an infection that is recalcitrant to therapy with an otherwise active agent. Finally, an antifungal agent cannot act if the patient does not take it in the prescribed manner. Noncompliance is a major cause of apparent “resistance” to antifungal therapy and additionally may contribute to the development of resistant strains.
The absorption, distribution, and metabolism of an antifungal agent all contribute to the effectiveness of the drug at the site of infection. Insufficient (too low) dosing practices may influence both therapeutic efficacy and the potential for resistance development (299). The ability of an antifungal agent to exhibit fungicidal activity versus fungistatic activity may be especially important in severe infections, in infections where the organism burden is high, and in the neutropenic host (64,300). Drug–drug interactions may also affect the activity of an antifungal agent: drugs that are metabolized by the cytochrome P450 enzyme system, such as rifamycin, may dramatically decrease the achievable concentrations of azole antifungal agents such as itraconazole and voriconazole (301).
Fungal properties, aside from the expression of known resistance factors, may also impact the clinical success or failure of antifungal therapy. The different morphologic forms of fungi (e.g., blastospore, hypha, pseudohypha, conidia, chlamydospore) may all have different susceptibilities to various antifungal agents (302,303). Phenotypic switching in Candida has been shown to have a dramatic impact on the susceptibility of various species to polyenes and azoles (223,304). Finally, the rate of growth of a fungus and whether or not it is growing in a planktonic or biofilm form can determine whether it will require low or high concentrations of an antifungal to inhibit growth (305,306). Candida spp are increasingly resistant to antifungals when grown in a biofilm compared to those grown in solution (307).
CONCLUSION
The incidence of infection with invasive mycoses continues to rise with the increasing immunosuppressed patient population. The recently expanded antifungal armamentarium offers the potential for more effective and less toxic therapy and these agents offer distinct pharmacologic profiles and indications for use. An understanding of the differing spectrum of activity, pharmacokinetics/pharmacodynamics, and dosing regimens enables the clinician to provide patients the best chance of favorable outcomes.
ANTIFUNGAL SUSCEPTIBILITY TESTING
The increasing number and diversity of invasive infections, expanding utilization of new and established antifungal agents, and recognition of antifungal resistance as an important clinical problem have contributed to the need for reproducible, clinically relevant antifungal susceptibility testing, especially for yeasts, but also for the filamentous fungi (298).
Rationale for Antifungal Susceptibility Testing
The central objective of all in vitro susceptibility testing is to help predict the likely impact of administration of the tested agent on the outcome of disease caused by the tested organism or similar organisms. As such, in vitro susceptibility tests of antifungal agents are performed for the same reasons as tests of antibacterial agents: (a) to provide a reliable estimate of the relative activities of antimicrobial agents, (b) to correlate with in vivo activity and predict the outcome of the therapy, (c) to provide a means by which to survey the development of resistance among a normally susceptible population of organisms, and (d) to predict the therapeutic potential of newly developed investigational agents. In the clinical microbiology/mycology laboratory, the focus of testing is on a specific isolate from an individual patient. In drug discovery, the focus of testing may be on the selection of the most potent of a series of compounds for further development. In antimicrobial resistance surveillance, the issue may be the tendency of resistance to emerge in initially susceptible isolates or species and to establish local, regional, or national patterns of resistance. In each of these settings, it is necessary to remember that outcome prediction is difficult and dependent on complex and dynamic biologic system and may substantially differ from results obtained in an artificial and well-defined matrix (antimicrobial susceptibility test). Decades of experience with antibacterial susceptibility testing confirms the limited degree of in vitro–in vivo correlation that can be achieved. The in vitro susceptibility of Enterococcus to trimethoprim-sulfamethoxazole and in vivo resistance to this combination is illustrative of this fact (308,309). In vitro susceptibility does not always predict successful therapy (298).
Development of Standardized Methods
At the present time, the state-of-the-art method for susceptibility testing of yeasts is comparable with that of bacteria (298). The CLSI Subcommittee on Antifungal Susceptibility Testing has developed and published approved methods for broth dilution testing of yeasts (310) and for disk diffusion testing of yeasts against fluconazole (311). These methods are reproducible, accurate, and available for use in clinical laboratories (312,313). Standardized methods have also been developed for broth dilution testing of filamentous fungi (314) but require further refinement and studies to establish the in vivo correlation with the in vitro data.
Standardized Broth Dilution Methods for Yeasts
In the United States, the National Committee for Clinical Laboratory Standards (NCCLS) (now CLSI) Subcommittee on Antifungal Susceptibility Testing was established in 1982 and focused on the key in vitro testing variables of inoculum preparation and size, medium composition, temperature and duration of incubation, and MIC end point determination in an effort to develop a standardized approach to antifungal susceptibility testing of yeasts (Candida spp and Cryptococcus neoformans) using a broth dilution format (315–319). As a result of these collaborative studies, consensus within the subcommittee was achieved on all of the variables, leading to the publication of a proposed broth macrodilution method, M27-P, in 1992 (320). This document was revised and published in 1995 as NCCLS document M27-T (tentative standard), which described the broth microdilution method and provided reference MIC ranges for two quality control (QC) strains for the available antifungal agents. In 1997, the subcommittee established interpretive MIC breakpoints for three antifungal agents (fluconazole, itraconazole, and 5-FC) (321) and the NCCLS-approved standard M27-A was published. Since then, the subcommittee has developed 24- and 48-hour reference QC MIC ranges for microdilution testing of both established (AmB, 5-FC, fluconazole, itraconazole, and ketoconazole) and newly introduced (voriconazole and posaconazole) agents (322). The results of these studies are included in the second edition of NCCLS document M27, M27-A2, published in 2002 (323). Since this time, the M27-A3 document was published in 2008 with the addition of caspofungin, micafungin, and anidulafungin susceptibility recommendations (324).
Adherence to the NCCLS M27-A3 method provides excellent intralaboratory and interlaboratory reproducibility and utilization of the recommended QC isolates will further ensure reliable test performance (324). The most recently published CLSI guidelines established revised breakpoints for fluconazole and voriconazole along with the echinocandins, which are species specific (Tables 6.4 and 6.5). These revised breakpoints recognize the wild-type distributions of Candida species against both azoles (fluconazole and voriconazole) and the echinocandins along with predicted pharmacokinetic and pharmacodynamic parameters of the drugs. These have generally resulted in much lower breakpoints for most species.
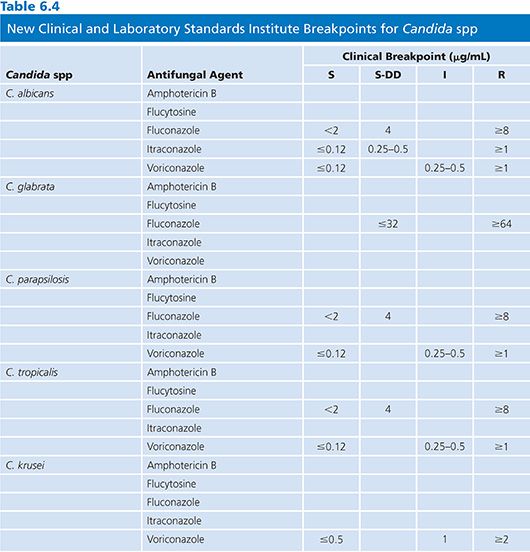
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


