Antifungal Agents
There are 200,000 known species of fungi, and estimates of the total size of Kingdom Fungi range to well over a million. Residents of the kingdom are quite diverse and include yeasts, molds, mushrooms, smuts, the pathogens Aspergillus fumigatus and Candida albicans, and the source of penicillin, Penicillium chrysogenum. Fortunately, only ~400 fungi cause disease in animals, and even fewer cause significant human disease. However, fungal infections are becoming more common in patients with compromised immune systems. Fungi are eukaryotes with unique cell walls containing glucans and chitin, and their eradication requires different strategies than those for treatment of bacterial infections. Available agents have effects on the synthesis of membrane and cell-wall components, on membrane permeability, on the synthesis of nucleic acids, and on microtubule/mitotic spindle function (Figure 57–1). Antifungal agents described in this chapter are discussed under two major headings, systemic and topical, although this distinction is somewhat arbitrary. The imidazole, triazole, and polyene antifungal agents may be used either systemically or topically, and many superficial mycoses can be treated either systemically or topically. Table 57–1 summarizes common mycoses and their pharmacotherapy.
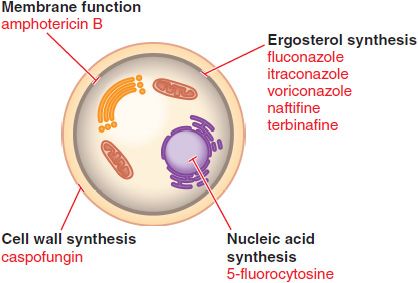
Figure 57–1 Sites of action of antifungal drugs. Amphotericin B and other polyenes (e.g., nystatin) bind to ergosterol in fungal cell membranes and increase membrane permeability. The imidazoles and triazoles (itraconazole, et al.) inhibit 14-α-sterol demethylase, prevent ergosterol synthesis, and lead to the accumulation of 14-α-methylsterols. The allylamines (e.g., naftifine and terbinafine) inhibit squalene epoxidase and prevent ergosterol synthesis. The echinocandins, such as caspofungin, inhibit the formation of glucans in the fungal cell wall.
Table 57–1
Pharmacotherapy of Mycoses
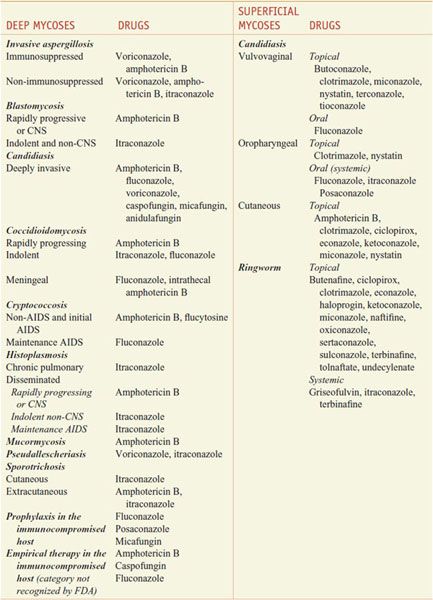
SYSTEMIC ANTIFUNGAL AGENTS
DRUGS FOR DEEPLY INVASIVE FUNGAL INFECTIONS
AMPHOTERICIN B. Amphotericin B is an amphoteric polyene macrolide with broad spectrum antifungal activity.
MECHANISM OF ACTION. Amphotericin B has useful clinical activity against a broad range of pathogenic fungi and limited activity against the protozoa Leishmania spp. and Naegleria fowleri. The antifungal activity of amphotericin B depends principally on its binding to a sterol moiety, primarily ergosterol in the membrane of sensitive fungi. By virtue of their interaction with these sterols, polyenes appear to form pores or channels that increase the permeability of the membrane, allowing leakage of a variety of small molecules (see Figure 57–1).
FORMULATIONS. Table 57–2 summarizes the pharmacokinetic properties of the 4 available preparations.
C-AMB (conventional amphotericin B, FUNGIZONE). Amphotericin B is insoluble in water but is formulated for intravenous infusion by complexing it with the bile salt deoxycholate. The complex is marketed as a lyophilized powder for injection. C-AMB forms a colloid in water, with particles largely <0.4 μm in diameter. Filters in intravenous infusion lines that trap particles >0.22 μm in diameter will remove significant amounts of drug. Addition of electrolytes to infusion solutions causes the colloid to aggregate.
ABCD. Amphotericin B colloidal dispersion (AMPHOTEC, AMPHOCIL) contains roughly equimolar amounts of amphotericin B and cholesteryl sulfate formulated for injection. Like C-AMB, ABCD forms a colloidal solution when dispersed in aqueous solution.
L-AMB. Liposomal amphotericin B is a small, unilamellar vesicle formulation of amphotericin B (AMBISOME). The drug is supplied as a lyophilized powder, which is reconstituted with sterile water for injection.
ABLC (ABELCET). Amphotericin B lipid complex is a complex of amphotericin B with lipids (dimyristoylphosphatidylcholine and dimyristoylphosphatidylglycerol).
The 3 lipid formulations collectively appear to reduce the risk of the patient’s serum creatinine doubling during therapy by 58%. However, the cost of the lipid formulations greatly exceeds that of C-AMB, making them unavailable in many countries.
ADME. GI absorption of all amphotericin B formulations is negligible and IV delivery is indicated for systemic use. Amphotericin B in plasma is more than 90% bound to proteins. Pharmacokinetic parameters vary with the formulation. Azotemia, liver failure, or hemodialysis does not have a measurable impact on plasma concentrations. Concentrations of amphotericin B (via C-AMB) in fluids from inflamed pleura, peritoneum, synovium, and aqueous humor are approximately two-thirds of trough concentrations in plasma. Little amphotericin B from any formulation penetrates into cerebrospinal fluid (CSF), vitreous humor, or normal amniotic fluid.
ANTIFUNGAL ACTIVITY; FUNGAL RESISTANCE. Amphotericin B has useful clinical activity against Candida spp., Cryptococcus neoformans, Blastomyces dermatitidis, Histoplasma capsulatum, Sporothrix schenckii, Coccidioides spp., Paracoccidioides braziliensis, Aspergillus spp., Penicillium marneffei, and the agents of mucormycosis. Amphotericin B has limited activity against the protozoa Leishmania spp. and N. fowleri. The drug has no antibacterial activity. Some isolates of Candida lusitaniae have been relatively resistant to amphotericin B. Aspergillus terreus and perhaps Aspergillus nidulans may be more resistant to amphotericin B than other Aspergillus species.
THERAPEUTIC USES. The recommended doses for each formulation are summarized in Table 57–2. Candida esophagitis responds to much lower doses than deeply invasive mycoses. Intrathecal infusion of C-AMB is useful in patients with meningitis caused by Coccidioides. Too little is known about intrathecal administration of lipid formulations to recommend them. C-AMB can be injected into the CSF of the lumbar spine, cisterna magna, or lateral cerebral ventricle. Fever and headache are common reactions that may be decreased by intrathecal administration of 10-15 mg of hydrocortisone. Local injections of amphotericin B into a joint or peritoneal dialysate fluid commonly produce irritation and pain. Intraocular injection following pars plana vitrectomy has been used successfully for fungal endophthalmitis. Intravenous administration of amphotericin B is the treatment of choice for mucormycosis and is used for initial treatment of cryptococcal meningitis, severe or rapidly progressing histoplasmosis, blastomycosis, coccidioidomycosis, and penicilliosis marneffei, as well as in patients not responding to azole therapy of invasive aspergillosis, extracutaneous sporotrichosis, fusariosis, alternariosis, and trichosporonosis. Amphotericin B (C-AMB or L-AMB) is often given to selected patients with profound neutropenia who have fever that does not respond to broad-spectrum antibacterial agents over 5-7 days.
Table 57–2
Pharmocokinetic Parameters for Amphotericin B Formulations after Multiple Administrations in Humans
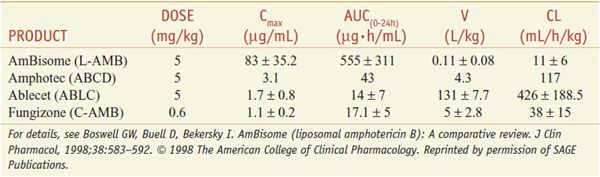
UNTOWARD EFFECTS. The major acute reactions to intravenous amphotericin B formulations are fever and chills. Infusion-related reactions are worst with ABCD and least with L-AMB. Tachypnea and respiratory stridor or modest hypotension also may occur, but true bronchospasm or anaphylaxis is rare. Patients with pre-existing cardiac or pulmonary disease may tolerate the metabolic demands of the reaction poorly and develop hypoxia or hypotension. The reaction ends spontaneously in 30-45 min; meperidine may shorten it. Pretreatment with oral acetaminophen or use of intravenous glucocorticoids at the start of the infusion decreases reactions.
Azotemia occurs in 80% of patients who receive C-AMB for deep mycoses. Lipid formulations are less nephrotoxic, being much less with ABLC, even less with L-AMB, and minimal with ABCD. Toxicity is dose-dependent and usually transient and increased by concurrent therapy with other nephrotoxic agents, such as aminoglycosides or cyclosporine. Permanent functional impairment is uncommon in adults with normal renal function prior to treatment unless the cumulative dose exceeds 3-4 g. Renal tubular acidosis and renal wasting of K+ and Mg2+ also may be seen during and for several weeks after therapy, often requiring repletion. Administration of 1 L of normal saline intravenously on the day that C-AMB is to be given has been recommended for adults who are able to tolerate the Na+ load.
Hypochromic, normocytic anemia commonly occurs during treatment with C-AMB. Anemia is less with lipid formulations and usually not seen over the first 2 weeks. The anemia is most likely due to decreased production of erythropoietin and often responds to administration of recombinant erythropoietin. Headache, nausea, vomiting, malaise, weight loss, and phlebitis at peripheral infusion sites are common. Arachnoiditis has been observed as a complication of injecting C-AMB into the CSF.
FLUCYTOSINE
Flucytosine (5-fluorocytosine) has a spectrum of antifungal activity that is considerably more restricted than that of amphotericin B.
MECHANISM OF ACTION. All susceptible fungi are capable of deaminating flucytosine to 5-fluorouracil (5-FU) (Figure 57–2), a potent antimetabolite that is used in cancer chemotherapy (see Chapter 61). Fluorouracil is metabolized first to 5-fluorouracil-ribose monophosphate (5-FUMP) by the enzyme uracil phosphoribosyl transferase (UPRTase). 5-FUMP then is either incorporated into RNA (via synthesis of 5-fluorouridine triphosphate) or metabolized to 5-fluoro-2′-deoxyuridine-5′-monophosphate (5-FdUMP), a potent inhibitor of thymidylate synthetase and thus of DNA synthesis. The selective action of flucytosine is due to the lack of cytosine deaminase in mammalian cells, which prevents metabolism to fluorouracil.
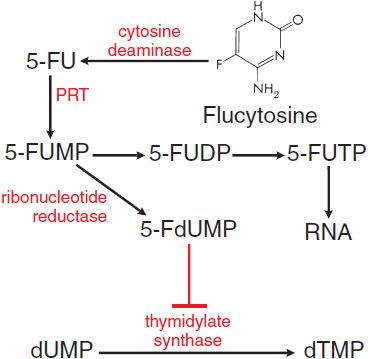
Figure 57–2 Action of flucytosine in fungi. Flucytosine (5-fluorocytosine) is transported by cytosine permease into the fungal cell, where it is deaminated to 5-fluorouracil (5-FU). The 5-FU is converted to 5-fluorouracil-ribose monophosphate (5-FUMP) and then is either converted to 5-fluorouridine triphosphate (5-FUTP) and incorporated into RNA or converted to 5-fluoro-2′-deoxyuridine-5′-monophosphate (5-FdUMP), a potent inhibitor of thymidylate synthase. 5-FUDP, 5-fluorouridine-5′-diphosphate; dUMP, deoxyuridine-5′-monophosphate; dTMP, deoxythymidine-5′-monophosphate; PRT, phosphoribosyltransferase.
ANTIFUNGAL ACTIVITY. Flucytosine has clinically useful activity against C. neoformans, Candida spp., and the agents of chromoblastomycosis.
FUNGAL RESISTANCE. Drug resistance arising during therapy (secondary resistance) is an important cause of therapeutic failure when flucytosine is used alone for cryptococcosis and candidiasis. The mechanism for this resistance can be loss of the permease necessary for cytosine transport or decreased activity of either UPRTase or cytosine deaminase (see Figure 57–2).
ADME. Flucytosine is absorbed rapidly and well from the GI tract and widely distributed in the body. Approximately 80% of a given dose is excreted unchanged in the urine. The t1/2 of the drug is 3-6 h but may reach 200 h in renal failure. The clearance of flucytosine is approximately equivalent to that of creatinine. Reduction of dosage is necessary in patients with decreased renal function, and concentrations of drug in plasma should be measured periodically (desirable range of peak concentrations, 50 and 100 μg/mL). Flucytosine is cleared by hemodialysis, and patients undergoing such treatment should receive a single dose of 37.5 mg/kg after dialysis; the drug also is removed by peritoneal dialysis.
THERAPEUTIC USES. Flucytosine (ANCOBON) is given orally at 50-150 mg/kg/day, in 4 divided doses at 6-h intervals. Flucytosine is used predominantly in combination with amphotericin B. An all-oral regimen of flucytosine plus fluconazole also has been advocated for therapy of AIDS patients with cryptococcosis, but the combination has substantial GI toxicity with no evidence that flucytosine adds benefit. The addition of flucytosine to ≥6 weeks of therapy with C-AMB runs the risk of substantial bone marrow suppression or colitis if the flucytosine dose is not promptly adjusted downward as amphotericin B–induced azotemia occurs. The guidelines for the treatment of cryptococcal meningoencephalitis recommend addition of flucytosine (100 mg/kg orally in 4 divided doses) for the first 2 weeks of treatment with amphotericin B in AIDS patients.
Untoward Effects. Flucytosine may depress the bone marrow and lead to leukopenia. Other untoward effects include rash, nausea, vomiting, diarrhea, and severe enterocolitis. In ~5% of patients, plasma levels of hepatic enzymes are elevated, but this effect reverses when therapy is stopped. Toxicity is more frequent in patients with AIDS or azotemia and when plasma drug concentrations exceed 100 μ/mL.
IMIDAZOLES AND TRIAZOLES
The azole antifungals include two broad classes, imidazoles and triazoles. Of the drugs now on the market in the U.S., clotrimazole, miconazole, ketoconazole, econazole, butoconazole, oxiconazole, sertaconazole, and sulconazole are imidazoles; terconazole, itraconazole, fluconazole, voriconazole, and posaconazole are triazoles. The topical use of azole antifungals is described in the second section of this chapter.
MECHANISM OF ACTION. The major effect of imidazoles and triazoles on fungi is inhibition of 14-α-sterol demethylase, a microsomal CYP (see Figure 57–1). Imidazoles and triazoles thus impair the biosynthesis of ergosterol for the cytoplasmic membrane and lead to the accumulation of 14-α-methylsterols. These methylsterols may disrupt the close packing of acyl chains of phospholipids, impairing the functions of certain membrane-bound enzyme systems, thus inhibiting growth of the fungi.
ANTIFUNGAL ACTIVITY. Azoles as a group have clinically useful activity against C. albicans, Candida tropicalis, Candida parapsilosis, Candida glabrata, C. neoformans, B. dermatitidis, H. capsulatum, Coccidioides spp., Paracoccidioides brasiliensis, and ringworm fungi (dermatophytes). Aspergillus spp., Scedosporium apiospermum (Pseudallescheria boydii), Fusarium, and S. schenckii are intermediate in susceptibility. Candida krusei and the agents of mucormycosis are more resistant. These drugs have antiprotozoal effects against Leishmania major. Posaconazole has slightly improved activity in vitro against the agents of mucormycosis.
RESISTANCE. Azole resistance emerges gradually during prolonged azole therapy, causing clinical failure in patients with far-advanced HIV infection and oropharyngeal or esophageal candidiasis. The primary mechanism of resistance in C. albicans is accumulation of mutations in ERG11, the gene coding for the 14-,-sterol demethylase; cross-resistance is conferred to all azoles.
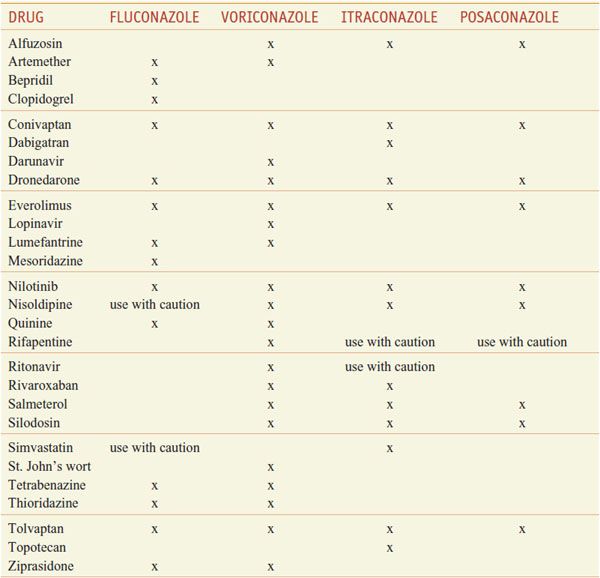
INTERACTION OF AZOLE ANTI-FUNGALS WITH OTHER DRUGS. The azoles interact with hepatic CYPs as substrates and inhibitors (Table 57–3), providing myriad possibilities for the interaction of azoles with many other medications. Thus, azoles can elevate plasma levels of some coadministered drugs (Table 57–4). Other coadministered drugs can decrease plasma concentrations of azole antifungal agents (Table 57–5). As a consequence of myriad interactions, combinations of certain drugs with azole antifungal medications may be contraindicated (Table 57–6).
Table 57–3
Interaction of Azole Antifungal Agents with Hepatic CYPs

Table 57–4
Drugs Exhibiting Elevated Plasma Concentrations When Co-Administered with Azole Anti-Fungal Agents
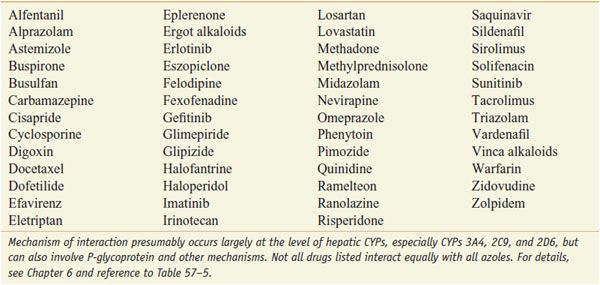
Table 57–5
Some Agents that Decrease Triazole Concentration
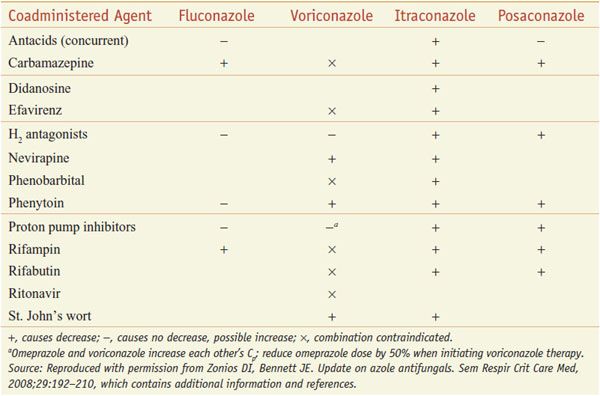
Table 57–6
Some Additional Contraindicated Azole Drug Combinations
KETOCONAZOLE
Ketoconazole, administered orally, has been replaced by itraconazole for the treatment of all mycoses except when the lower cost of ketoconazole outweighs the advantage of itraconazole. Ketoconazole sometimes is used to inhibit excessive production of glucocorticoids in patients with Cushing syndrome (see Chapter 42) and is available for topical use.
ITRACONAZOLE
Itraconazole lacks ketoconazole’s corticosteroid suppression while retaining most of ketoconazole’s pharmacological properties and expanding the antifungal spectrum. This synthetic triazole is an equimolar racemic mixture of 4 diastereoisomers.
ADME. Itraconazole (SPORANOX, others) is available as a capsule and a solution in hydroxypropyl-β-cyclodextrin for oral use. The capsule form of the drug is best absorbed in the fed state, but the oral solution is better absorbed in the fasting state, providing peak plasma concentrations >150% of those obtained with the capsule. Itraconazole is metabolized in the liver; it is both a substrate for and a potent inhibitor of CYP3A4. Itraconazole is present in plasma with an approximately equal concentration of a biologically active metabolite, hydroxy-itraconazole. The native drug and metabolite are >99% bound to plasma proteins. Neither appears in urine or CSF. The t1/2 of itraconazole at steady state is ~30-40 h. Steady-state levels of itraconazole are not reached for 4 days and those of hydroxy-itraconazole for 7 days; thus, loading doses are recommended when treating deep mycoses. Severe liver disease will increase itraconazole plasma concentrations, but azotemia and hemodialysis have no effect.
DRUG INTERACTIONS. Tables 57–4, 57–5, and 57–6 list select interactions of azoles with other drugs.
THERAPEUTIC USES. Itraconazole is the drug of choice for patients with indolent, nonmeningeal infections due to B. dermatitidis, H. capsulatum, P. brasiliensis, and Coccidioides immitis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


