





 , Kidrolase
, Kidrolase 
Mechanisms of Cytotoxic Action
Table 82.2 shows the principal mechanisms by which the cytotoxic anticancer drugs act. As the table shows, most cytotoxic agents disrupt processes related to synthesis of DNA or its precursors. In addition, some agents (e.g., vinblastine, vincristine) act specifically to block mitosis, and one drug—asparaginase—disrupts synthesis of proteins. Note that, with the exception of asparaginase, all of the cytotoxic drugs disrupt processes carried out exclusively by cells that are undergoing replication. As a result, these drugs are most toxic to tissues that have a high growth fraction (i.e., a high proportion of proliferating cells).
Cell-Cycle Phase Specificity
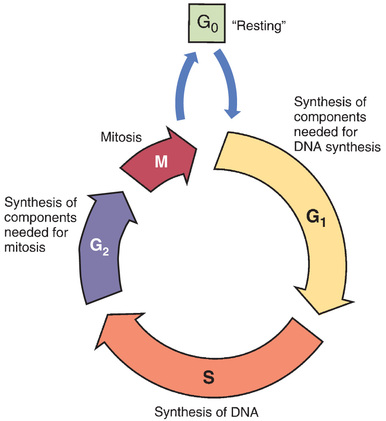
Some anticancer agents, known as cell-cycle phase–specific drugs, are effective only during a specific phase of the cell cycle. (See Box 82.1.) Other anticancer agents, known as cell-cycle phase–nonspecific drugs, can affect cells during any phase of the cell cycle. About half of the cytotoxic anticancer drugs are phase specific, and the other half are phase nonspecific. The phase specificity of individual cytotoxic agents is shown in Table 82.2.
Cell-Cycle Phase–Specific Drugs
Phase-specific agents are toxic only to cells that are passing through a particular phase of the cell cycle (see Fig. 82.1). Vincristine, for example, acts by causing mitotic arrest and hence is effective only during M phase. Other agents act by disrupting DNA synthesis and hence are effective only during S phase. Because of their phase specificity, these drugs are toxic only to cells that are active participants in the cell cycle; cells that are “resting” in G0 will not be harmed. Obviously, if these drugs are to be effective, they must be present as neoplastic cells cycle through the specific phase in which they act. Accordingly, these drugs must be present for an extended time. To accomplish this, phase-specific drugs are often administered by prolonged infusion. Alternatively, they can be given in multiple doses at short intervals over an extended time. Because the dosing schedule is so critical to therapeutic response, phase-specific drugs are also known as schedule-dependent drugs.
Cell-Cycle Phase–Nonspecific Drugs
The phase-nonspecific drugs can act during any phase of the cell cycle, including G0. Among the phase-nonspecific drugs are the alkylating agents and most antitumor antibiotics. Because phase-nonspecific drugs can injure cells throughout the cell cycle, whereas phase-specific drugs cannot, phase-nonspecific drugs can increase cell kill when combined with phase-specific drugs.
Although the phase-nonspecific drugs can cause biochemical lesions at any time during the cell cycle, as a rule these drugs are more toxic to proliferating cells than to cells in G0. There are two reasons why this is so. First, cells in G0 have time to repair drug-induced damage before it can result in significant harm. In contrast, proliferating cells often lack time for repair. Second, toxicity may not become manifest until the cells attempt to proliferate. For example, many alkylating agents act by producing cross-links between DNA strands. Although these biochemical lesions can be made at any time, they are largely without effect until cells attempt to replicate DNA. This is much like inflicting a flat tire on an automobile: the tire can be deflated at any time; however, loss of air is consequential only if the car is moving. Carrying the analogy further, if the flat occurs while the car is stopped, and is repaired before travel is attempted, the flat will have no functional effect at all.
It should be noted that some dividing cells are more vulnerable than others. Specifically, cells that divide frequently are harmed more readily than cells that divide infrequently because the cytotoxic drugs have more opportunities to act.
Toxicity to Normal Cells
Toxicity to normal cells is a major barrier to successful chemotherapy. Injury to normal cells occurs primarily in tissues where the growth fraction is high: bone marrow, gastrointestinal (GI) epithelium, hair follicles, and germinal epithelium of the testes. Drug-induced injury to each of these tissues is discussed in detail later. For now, let’s consider injury to normal cells as a group.
Toxicity to normal cells is dose limiting. That is, dosage cannot exceed an amount that produces the maximally tolerated injury to normal cells. Although very large doses of cytotoxic drugs might be able to produce cure, these doses cannot be given because they are likely to kill the patient.
Why are cytotoxic anticancer drugs so harmful to normal tissues? Because these drugs lack selective toxicity. That is, they cannot kill target cells without also killing other cells with which the target cells are in intimate contact. We encountered this concept in Chapter 68. As noted there, successful antimicrobial therapy is possible because antimicrobial drugs are highly selective in their toxicity. Penicillin, for example, can readily kill invading bacteria while being virtually harmless to cells of the host. This high degree of selective toxicity stands in sharp contrast to the lack of selectivity displayed by cytotoxic anticancer drugs.
Why have we been unable to develop drugs that selectively kill neoplastic cells? Because neoplastic cells and normal cells are very similar: differences between them are quantitative rather than qualitative. To make a cytotoxic drug that is truly selective, the target cell must have a biochemical feature that normal cells lack. Unfortunately, we have yet to identify unique biochemical features that would render cancer cells vulnerable to selective attack. Nevertheless, there is reason for hope: our expanding knowledge of cancer biology is revealing potential new targets for anticancer drugs. Exploiting these targets may lead to anticancer drugs that are more selective than the drugs we have now.
Major Toxicities of Chemotherapeutic Drugs
The agents used for cancer chemotherapy constitute our most toxic group of medicines. In the discussion that follows, we consider the more common toxicities of the cytotoxic anticancer drugs along with steps that can be taken to minimize harm and discomfort.
Bone Marrow Suppression
Chemotherapeutic drugs are highly toxic to the bone marrow, a tissue with a high proportion of proliferating cells. Myelosuppression reduces the number of circulating neutrophils, platelets, and erythrocytes. Loss of these cells has three major consequences: (1) infection (from loss of neutrophils); (2) bleeding (from loss of platelets); and (3) anemia (from loss of erythrocytes).
Neutropenia
Neutrophils (neutrophilic granulocytes) are white blood cells that play a critical role in fighting infection. In patients with neutropenia (a reduction in circulating neutrophils), both the incidence and severity of infection are increased. Infections that are normally benign (e.g., candidiasis) can become life-threatening. Infection secondary to neutropenia is one of the most serious complications of chemotherapy.
With most anticancer drugs, onset of neutropenia is rapid and recovery develops relatively quickly. Neutropenia begins to develop a few days after dosing, and the lowest neutrophil count, called the nadir, occurs between days 10 and 14. Neutrophil counts then recover a week or so later. Patients are at highest risk during the nadir. Accordingly, special care should be taken to prevent infection.
With some anticancer drugs, neutropenia is delayed. Neutrophil counts begin to fall in 1 to 2 weeks and reach their nadir between weeks 3 and 4. Full recovery may not occur until after week 7.
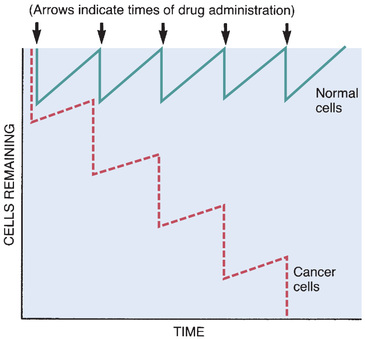
Neutrophil counts must be monitored. Normal counts range from 2500 to 7000 cells/mm3. If neutropenia is substantial (absolute neutrophil count below 500/mm3), chemotherapy should be withheld until neutrophil counts return toward normal. Fortunately, normal cells such as neutrophils repopulate faster than malignant cells (see Fig. 82.2).
Lack of neutrophils confounds the diagnosis of infection. Why? Because the usual signs of infection (e.g., pus, abscesses, infiltrates on the chest radiograph) depend on neutrophils being present. In the absence of neutrophils, fever is the principal early sign of infection.
Patients must be their own first line of defense against infection. They should be made aware of their elevated risk for infection and taught how to minimize contagion. They should be informed that fever may be the only indication of infection and instructed to report immediately if fever develops. Because infection is commonly acquired through contact with other people, hospitalized patients should be instructed to refuse direct contact with anyone who has not washed his or her hands in the patient’s presence. This rule applies not only to visiting friends and relatives but also to nurses, physicians, and all other hospital staff. The normal flora of the body is a major source of infection; the risk for acquiring an infection with these microbes can be reduced by daily examination and cleansing of the skin and oral cavity.
Hospitalization of the infection-free neutropenic patient is controversial. Some providers feel that hospitalization increases the risk for acquiring a serious infection. Why? Because hospitals harbor drug-resistant microbes, which can make hospital-acquired (nosocomial) infections especially difficult to treat. Accordingly, these providers recommend that neutropenic patients stay at home as long as they remain infection free.
If neutropenic patients are hospitalized, every precaution must be taken to prevent nosocomial infection. Patients should be given an isolation room and monitored frequently for fever. Certain foods (e.g., lettuce) abound in pathogenic bacteria and must be avoided.
When a neutropenic patient develops an infection, immediate and vigorous intervention is required. Specimens for culture should be taken to determine the identity and drug sensitivity of the infecting organism. While awaiting reports on the cultures, empiric therapy with intravenous (IV) antibiotics should be instituted. Initial therapy is usually done with a single drug active against Pseudomonas species and other gram-negative bacteria. Options include ceftazidime, imipenem, and doripenem. If the patient develops sepsis, an aminoglycoside (e.g., tobramycin, amikacin) is added. If the patient remains febrile, vancomycin is added (for gram-positive coverage).
Colony-stimulating factors can minimize neutropenia. Three preparations are available: granulocyte colony-stimulating factor (filgrastim), long-acting granulocyte colony-stimulating factor (pegfilgrastim), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (sargramostim). All three drugs act on the bone marrow to enhance granulocyte (neutrophil) production. Colony-stimulating factors can decrease the incidence, magnitude, and duration of neutropenia. As a result, they can decrease the incidence and severity of infection as well as the need for IV antibiotics and hospitalization.
Thrombocytopenia
Bone marrow suppression can cause thrombocytopenia (a reduction in circulating platelets), thereby increasing the risk for serious bleeding. Bleeding from the nose and gums is relatively common. Bleeding from the gums can be reduced by avoiding vigorous tooth brushing. Drugs that promote bleeding (e.g., aspirin, anticoagulants) should not be used. When a mild analgesic is required, acetaminophen, which does not promote bleeding, is preferred to aspirin. For patients with severe thrombocytopenia, platelet infusions are the mainstay of treatment. Platelet production can be stimulated with oprelvekin [Neumega]. However, owing to limited efficacy and flu-like reactions, oprelvekin is not often used.
Caution should be exercised when performing procedures that might promote bleeding. IV needles should be inserted with special care, and intramuscular (IM) injections should be avoided. Blood pressure cuffs should be applied cautiously because overinflation may cause bruising or bleeding.
Anemia
Anemia is defined as a reduction in the number of circulating erythrocytes (red blood cells). Although anticancer drugs can suppress erythrocyte production, anemia is much less common than neutropenia or thrombocytopenia. Why? Because circulating erythrocytes have a long life span (120 days), which usually allows erythrocyte production to recover before levels of existing erythrocytes fall too low.
If anemia does develop, it can be treated with a transfusion or with erythropoietin (epoetin alfa or darbepoetin alfa), a hormone that stimulates production of red blood cells. Because transfusions require hospitalization, whereas epoetin can be administered at home, epoetin therapy can spare the patient inconvenience. However, erythropoietin has two huge drawbacks. First, it cannot be used in patients with leukemias and other myeloid malignancies (because it can stimulate proliferation of these cancers). Second, it shortens survival in all cancer patients and hence is indicated only when the treatment goal is palliation. Clearly, erythropoietin should not be used when the goal is cure or prolongation of life. The basic pharmacology of erythropoietin is discussed in Chapter 45.
Digestive Tract Injury
The epithelial lining of the GI tract has a very high growth fraction, so it is exquisitely sensitive to cytotoxic drugs. Stomatitis and diarrhea are common. Severe GI injury can be life-threatening.
Stomatitis
Stomatitis (inflammation of the oral mucosa) often develops a few days after the onset of chemotherapy and may persist for 2 or more weeks after treatment has ceased. Inflammation can progress to denudation and ulceration and is often complicated by infection. Pain can be severe, inhibiting eating, speaking, and swallowing. Management includes good oral hygiene and a bland diet. Topical antifungal drugs may be needed to control infection with Candida albicans. For patients with mild stomatitis, pain can be managed with a mouthwash containing a topical anesthetic (e.g., lidocaine) plus an antihistamine (e.g., diphenhydramine). For patients with severe stomatitis, a systemic opioid is needed for pain. In some cases, stomatitis is so severe that chemotherapy has to be interrupted. For patients being treated for hematologic malignancies, palifermin [Kepivance], a chemoprotective agent and keratinocyte growth factor, can decrease the severity of stomatitis.
Diarrhea
By injuring the epithelial lining of the intestine, anticancer drugs can impair absorption of fluids and other nutrients, thereby causing diarrhea. Diarrhea can be reduced with oral loperamide, a nonabsorbable opioid that slows gut motility by activating local opioid receptors.
Nausea and Vomiting
Nausea and vomiting are common sequelae of cancer chemotherapy. These responses, which result in part from direct stimulation of the chemoreceptor trigger zone, can be both immediate and dramatic and may persist for hours or even days. In some cases, discomfort is so great as to prompt refusal of further treatment.
You should appreciate that nausea and vomiting associated with chemotherapy are much more severe than with other medications. Whereas these reactions are generally unremarkable with most drugs, they must be considered major and characteristic toxicities of cytotoxic drugs. The emetogenic potential of several intravenous agents is shown in Table 82.3.
TABLE 82.3
Emetogenic Potential of Selected Intravenous Anticancer Drugs
| Severe | |
| Moderate | |
| Low | |
| Minimal |
Nausea and vomiting can be reduced by premedication with antiemetics. These drugs offer three benefits: (1) reduction of anticipatory nausea and vomiting, (2) prevention of dehydration and malnutrition secondary to frequent nausea and vomiting, and (3) promotion of compliance with chemotherapy by reducing discomfort. Combinations of antiemetics are more effective than single-drug therapy. The regimen of choice for patients taking highly emetogenic drugs consists of aprepitant [Emend], dexamethasone, and a serotonin antagonist, such as ondansetron [Zofran]. The use of antiemetics for chemotherapy-induced nausea and vomiting is discussed in Chapter 64.
Other Important Toxicities
Alopecia
Reversible alopecia (hair loss) results from injury to hair follicles. Alopecia can occur with most cytotoxic anticancer drugs. Hair loss begins 7 to 10 days after the onset of treatment and reaches maximal loss in 1 to 2 months. Regeneration begins 1 to 2 months after the last course of treatment.
Although alopecia is not dangerous, it is nonetheless very upsetting. In fact, for many cancer patients, alopecia is second only to vomiting as their greatest treatment-related fear. If drugs are expected to cause hair loss, the patient should be forewarned. For patients who choose to wear a hairpiece or wig, one should be selected before hair loss occurs. Hairpieces are tax deductible as medical expenses and are covered by some insurance plans.
To some degree, hair loss can be prevented by cooling the scalp while chemotherapy is being administered. Cooling causes vasoconstriction and thereby reduces drug delivery to hair follicles. Unfortunately, scalp cooling is uncomfortable, causes headache, and creates a small risk for cancer recurrence in the scalp (because drug delivery is reduced).
Reproductive Toxicity
The developing fetus and the germinal epithelium of the testes have high growth fractions. As a result, both are highly susceptible to injury by cytotoxic drugs, especially the alkylating agents. These drugs can interfere with embryogenesis, causing death of the early embryo. They may also cause fetal malformation. Risk is highest during the first trimester, and hence chemotherapy should generally be avoided during this time. However, after 18 weeks of gestation, risk appears to be very low: according to a 2012 report in Lancet, exposure during this time does not cause neurologic, cardiac, or any other fetal abnormalities. Drug effects on the ovaries may result in amenorrhea, menopausal symptoms, and atrophy of the vaginal epithelium.
Cytotoxic drugs can cause irreversible sterility in males. Men should be forewarned and counseled about sperm banking.
Hyperuricemia
Hyperuricemia is defined as an excessive level of uric acid in the blood. Uric acid, a compound with low solubility, is formed by the breakdown of DNA after cell death. Hyperuricemia is especially common after treatment for leukemias and lymphomas (because therapy results in massive cell kill). The major concern with hyperuricemia is injury to the kidneys secondary to deposition of uric acid crystals in renal tubules. The risk for crystal formation can be reduced by increasing fluid intake. In patients with leukemias and lymphomas, in whom hyperuricemia is likely, prophylaxis with allopurinol is the standard of care. (As discussed in Chapter 58, allopurinol prevents hyperuricemia by inhibiting xanthine oxidase, an enzyme involved in converting nucleic acids to uric acid.) If hyperuricemia develops despite use of allopurinol, it can be managed with rasburicase, an enzyme that catalyzes uric acid degradation).
Local Injury From Extravasation of Vesicants
Certain anticancer drugs, known as vesicants, are highly chemically reactive. These drugs can cause severe local injury if they make direct contact with tissues. Vesicants are administered intravenously, usually into a central line (because rapid dilution in venous blood minimizes the risk for injury). When a peripheral line is used, administration is by IV push into a freely flowing IV line. Sites of previous irradiation should be avoided. Extreme care must be exercised to prevent extravasation because leakage can produce high local concentrations, resulting in prolonged pain, infection, and loss of mobility. Severe injury can lead to necrosis and sloughing, requiring surgical débridement and skin grafting. If extravasation occurs, the infusion should be stopped immediately. Because of the potential for severe tissue damage, vesicants should be administered only by clinicians specially trained to handle them safely.
Unique Toxicities
In addition to the toxicities discussed previously, which generally apply to the cytotoxic drugs as a group, some agents produce unique toxicities. For example, a number of drugs can cause peripheral sensory neuropathy, manifesting as numbness or tingling in the fingers and toes and around the mouth and throat. Neuropathy may impede activities of daily living, such as buttoning clothing, writing, or just holding things. Anthracyclines such as daunorubicin and doxorubicin can cause serious injury to the heart. Ifosfamide, a nitrogen mustard, can cause severe hemorrhagic cystitis.
Carcinogenesis
Along with their other adverse actions, anticancer drugs have one final and ironic toxicity: these drugs, which are used to treat cancer, have caused cancer in some patients. Cancer results from drug-induced damage to DNA and is most likely to occur with alkylating agents. Cancers caused by anticancer drugs may take many years to appear and are hard to treat.
Making the Decision to Treat
From the preceding discussion of toxicities, it is clear that cytotoxic anticancer drugs can cause great harm. Given the known dangers of these drugs, we must ask why such toxic substances are given to sick people at all. The answer lies with the primary rule of therapeutics, which states that the benefits of treatment must outweigh the risks. For most patients undergoing chemotherapy, the conditions of this rule are met. That is, although the toxicities of the anticancer drugs can be significant, the potential benefits (cure, prolonged life, palliation) justify the risks. However, the desirability of treating cancer with drugs is not always obvious. There are patients whose chances of being helped by chemotherapy are remote, whereas the risk for serious toxicity is high. Because the potential benefits for some patients are small and the risks are large, the decision to institute chemotherapy must be made with care.
Before a decision to treat can be made, the patient must be given some idea of the benefits the proposed therapy might offer. Three basic benefits are possible: cure, prolongation of life, and palliation. For treatment to be justified, there should be reason to believe that at least one of these benefits will be forthcoming. If a patient cannot be offered some reasonable hope of cure, prolonged life, or palliation, it would be difficult to justify treatment.
The most important factors for predicting the outcome of chemotherapy are (1) the general health of the patient and (2) the responsiveness of the type of cancer the patient has. General health status is assessed by measuring performance status, frequently using the Karnofsky Performance Scale (Table 82.4). A Karnofsky score of less than 40 indicates the patient is debilitated and not likely to tolerate the additional stress of chemotherapy. Accordingly, patients with a low Karnofsky rating should not receive anticancer drugs—unless their cancer is known to be especially responsive.
TABLE 82.4
Karnofsky Performance Scale
| Definition | Percentage | Criteria |
| Able to carry on normal activity and work; no special care needed | 100 | Normal; no complaints; no evidence of disease |
| 90 | Able to carry on normal activity; minor signs or symptoms of disease | |
| 80 | Normal activity with effort; some signs or symptoms of disease | |
| Unable to work; able to live at home and care for most personal needs; a varying amount of assistance needed | 70 | Cares for self; unable to carry on normal activity or do active work |
| 60 | Requires occasional assistance; able to care for most needs | |
| 50 | Requires considerable assistance and frequent medical care | |
| Unable to care for self; requires equivalent of institutional or hospital care; disease may be progressing rapidly | 40 | Disabled; requires special care and assistance |
| 30 | Severely disabled; hospitalization is indicated although death not imminent | |
| 20 | Very sick, hospitalization necessary; active supportive treatment necessary | |
| 10 | Moribund; fatal processes progressing rapidly | |
| 0 | Dead |
The responsiveness of specific cancers is not highly predictable: some patients with a specific type of cancer may respond well, but others may not. Nonetheless, we should still try to assess whether treatment is likely to produce cure, palliation, or prolonged life. If a positive outcome is deemed likely, the patient should almost always be treated, even if his or her Karnofsky score is low. In contrast, if a positive outcome is deemed highly unlikely, the patient should be treated only after careful consideration, so as to avoid the discomforts of a course of treatment that has little to offer.
An important requirement for deciding in favor of chemotherapy is that the effect of treatment be measurable. That is, there must be some objective means of determining the cancer’s response to drugs. For solid tumors, we should be able to measure a decrease in tumor size (or at least inhibition of further growth). For hematologic cancers, we should be able to measure a decrease in neoplastic cells in blood and bone marrow. If we have no way to measure the response of a cancer, then we have no way of knowing if treatment has done any good. If we cannot determine that drugs are doing something beneficial, there is little justification for giving them.
Clearly, not all patients are candidates for chemotherapy. The decision to institute treatment must be individualized. Patients should be informed as accurately as possible about the potential risks and benefits of the proposed therapy. When the decision to treat is made, it should be the result of collaboration between the patient, family, and physician and should reflect a conviction on the part of the patient that, within his or her set of values, the potential benefits outweigh the inherent risks.
Cytotoxic Agents
Alkylating Agents
The family of alkylating agents consists of nitrogen mustards, nitrosoureas, and other compounds. The alkylating agents are shown in Table 82.2.
Shared Properties
Mechanism of Action
The alkylating agents are highly reactive compounds that can transfer an alkyl group to various cell constituents. Cell kill results primarily from alkylation of DNA. As a rule, alkylating agents interact with DNA by forming a covalent bond with a specific nitrogen atom in guanine.
Some alkylating agents have two reactive sites, whereas others have only one. Alkylating agents with two reactive sites (bifunctional agents) are able to bind DNA in two places to form cross-links. These bridges may be formed within a single DNA strand or between parallel DNA strands. Fig. 82.3 shows the production of interstrand cross-links by nitrogen mustard. Alkylating agents with only one reactive site (monofunctional agents) lack the ability to form cross-links but can still bind to a single guanine in DNA.
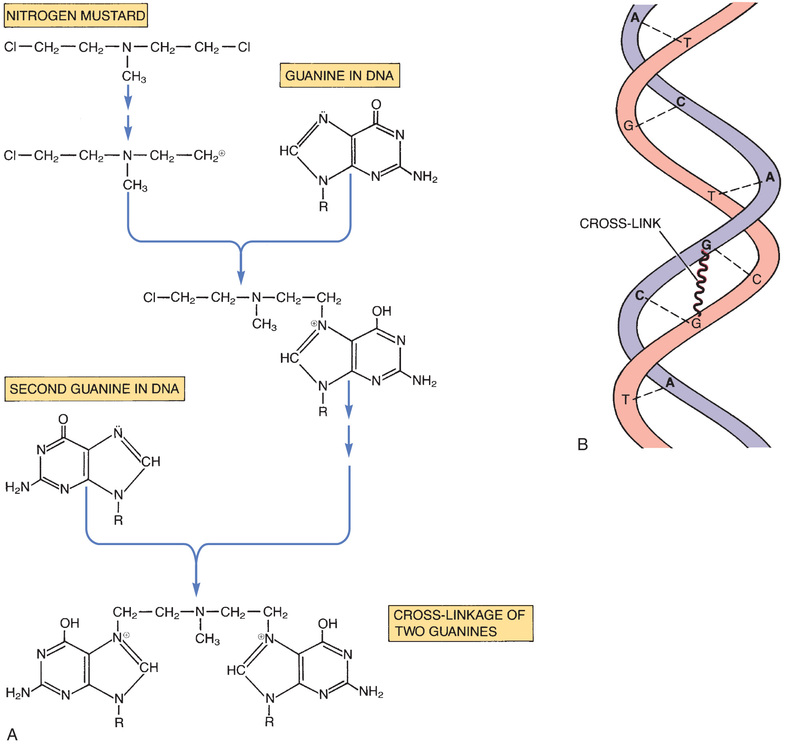
The consequences of guanine alkylation are miscoding, scission of DNA strands, and, if cross-links have been formed, inhibition of DNA replication. Because cross-linking of DNA is especially injurious, cell death is more likely with bifunctional agents than with monofunctional agents.
Because alkylation reactions can take place at any time during the cell cycle, alkylating agents are considered cell-cycle phase nonspecific. However, most of these drugs are more toxic to dividing cells—especially cells that divide rapidly—than they are to cells in G0. Why? Because (1) alkylation of DNA produces its most detrimental effects when cells attempt to replicate DNA and (2) quiescent cells are often able to repair damage to DNA before it can affect cell function.
Toxicities
Alkylating agents are toxic to tissues that have a high growth fraction. Accordingly, these drugs may injure cells of the bone marrow, hair follicles, GI mucosa, and germinal epithelium. Blood dyscrasias—neutropenia, thrombocytopenia, and anemia—caused by bone marrow suppression are of greatest concern. Nausea and vomiting occur with all alkylating agents. Also, several of these drugs are vesicants and hence must be administered through a free-flowing IV line. The major dose-limiting toxicities of individual drugs are provided in Table 82.2.
Nitrosoureas
The nitrosoureas are bifunctional alkylating agents and are active against a broad spectrum of neoplastic diseases. Cell kill results from cross-linking DNA. Unlike many anticancer drugs, the nitrosoureas are highly lipophilic and hence can readily penetrate the blood-brain barrier. As a result, these drugs are especially useful against cancers of the CNS. The major dose-limiting toxicity is delayed bone marrow suppression.
Platinum Compounds
The platinum-containing anticancer drugs—cisplatin, carboplatin, and oxaliplatin—are similar to the alkylating agents and are often classified as such. Like the bifunctional alkylating agents, the platinum compounds produce cross-links in DNA and hence are cell-cycle phase nonspecific.
Antimetabolites
Antimetabolites are structural analogs of important natural metabolites. Because they resemble natural metabolites, these drugs are able to disrupt critical metabolic processes. Some antimetabolites inhibit enzymes that synthesize essential cellular constituents. Others undergo incorporation into DNA and thereby disrupt DNA replication and function.
Antimetabolites are effective only against cells that are active participants in the cell cycle. Most antimetabolites are S-phase specific, although some can act during any phase of the cycle, except G0. To be effective, agents that are S-phase specific must be present for an extended time.
There are three classes of antimetabolites: (1) folic acid analogs, (2) pyrimidine analogs, and (3) purine analogs. Members of each class, along with their dose-limiting toxicities, are shown in Table 82.2.
Folic Acid Analogs
Folic acid, in its active form, is needed for several essential biochemical reactions. The folic acid analogs block the conversion of folic acid to its active form. At this time, three analogs of folic acid are used against cancer: methotrexate, pemetrexed, and pralatrexate. Other folate analogs are used to treat bacterial infections (trimethoprim), malaria (pyrimethamine), and Pneumocystis jiroveci pneumonia (trimetrexate).
Pyrimidine Analogs
The pyrimidine analogs are cytarabine, fluorouracil, capecitabine, floxuridine, and gemcitabine.
Pyrimidines—cytosine, thymine, and uracil—are bases employed in the biosynthesis of DNA and RNA. The pyrimidine analogs, because of their structural similarity to naturally occurring pyrimidines, can act in several ways: (1) they can inhibit biosynthesis of pyrimidines, (2) they can inhibit biosynthesis of DNA and RNA, and (3) they can undergo incorporation into DNA and RNA and thereby disrupt nucleic acid function. All of the pyrimidine analogs are prodrugs that must be converted to their active forms in the body.
Purine Analogs
The purine analogs are cladribine, clofarabine, fludarabine, mercaptopurine, nelarabine, pentostatin, and thioguanine. Like the pyrimidines, the purines—adenine, guanine, and hypoxanthine—are bases employed for biosynthesis of nucleic acids. Purine analogs discussed in other chapters are used for immunosuppression, antiviral therapy, and gout.
Hypomethylating Agents
Hypomethylating agents are so-called because they inhibit DNA methyltransferase, an enzyme that puts methyl groups onto DNA components. Drugs in this category include azacitidine and decitabine. Both are analogs of cytidine, a component of RNA.
Antitumor Antibiotics
The antitumor antibiotics are cytotoxic drugs originally isolated from cultures of Streptomyces species. They fall into two major groups: anthracyclines and nonanthracyclines. Antitumor antibiotics are used only to treat cancer; they are not used to treat infections. All of these drugs injure cells through direct interaction with DNA.
Anthracyclines
Five of the antitumor antibiotics are derivatives of anthracycline: doxorubicin (conventional and liposomal), daunorubicin (conventional and liposomal), epirubicin, idarubicin, and valrubicin. A sixth drug, mitoxantrone, is often categorized as an anthracycline because of its close similarity to drugs in this category. All can cause severe bone marrow suppression and heart damage. In some patients, cardiotoxicity has led to fatal heart failure.
Nonanthracyclines
There are three nonanthracycline antitumor antibiotics: dactinomycin, bleomycin, and mitomycin. In contrast to anthracyclines, nonanthracyclines do not injure the heart. However, these drugs do have serious toxicities of their own. Table 82.2 details dose-limiting toxicities of each drug.
Mitotic Inhibitors
Mitotic inhibitors are drugs that act during M phase to prevent cell division. There are two major groups of these drugs—vinca alkaloids and taxanes—as well as three other drugs that belong to neither group. Of these, vincristine is unique in that it spares the bone marrow and thus causes very little damage with regard to hematopoiesis.
Topoisomerase Inhibitors
Topoisomerases are nuclear enzymes that alter the shape (topology) of supercoiled DNA. Without the actions of topoisomerases, the double helix would be too tangled to permit DNA replication, RNA synthesis, or DNA repair. How do topoisomerases alter DNA configuration? They make a cut in the DNA strand—which permits the strand to relax in the vicinity of the cut—and then later they reseal the cut. There are two types of topoisomerase, known as topoisomerase I and topoisomerase II. Topoisomerase I makes single-strand cuts, and topoisomerase II makes double-strand cuts. Of the four topoisomerase inhibitors in current use, two—topotecan and irinotecan—inhibit topoisomerase I, and the other two—etoposide and teniposide—inhibit topoisomerase II. The actions of these drugs are partly like those of the antitumor antibiotics discussed previously, which inhibit topoisomerase II and intercalate DNA.
Miscellaneous Cytotoxic Drugs
Some cytotoxic drugs are in categories of their own. A few examples follow.
Asparaginase
Asparaginase [Elspar, Erwinase  , Kidrolase
, Kidrolase  ] is an enzyme that converts asparagine, an essential amino acid, into aspartic acid. By converting asparagine to aspartic acid, the drug deprives cells of asparagine needed to synthesize proteins. However, not all cells are affected. In fact, toxicity from asparaginase is limited almost exclusively to leukemic lymphoblasts because these cells are unable to manufacture their own asparagine like normal cells can. Therefore normal cells are able to replace the asparagine that asparaginase took away, but leukemic lymphoblasts can’t. Asparaginase appears to act selectively during G1.
] is an enzyme that converts asparagine, an essential amino acid, into aspartic acid. By converting asparagine to aspartic acid, the drug deprives cells of asparagine needed to synthesize proteins. However, not all cells are affected. In fact, toxicity from asparaginase is limited almost exclusively to leukemic lymphoblasts because these cells are unable to manufacture their own asparagine like normal cells can. Therefore normal cells are able to replace the asparagine that asparaginase took away, but leukemic lymphoblasts can’t. Asparaginase appears to act selectively during G1.
Hydroxyurea
Hydroxyurea [Hydrea, Droxia] inhibits DNA replication by suppressing synthesis of DNA precursors. Specifically, the drug inhibits ribonucleoside diphosphate reductase, the enzyme that converts ribonucleotides into their corresponding deoxyribonucleotides. In the absence of deoxyribonucleotides, DNA cannot be made. Hydroxyurea is S-phase specific.
Mitotane
Mitotane [Lysodren] is a structural analog of two insecticides: DDD and DDT. For reasons that are not understood, the drug is selectively toxic to cells of the adrenal cortex, both normal and neoplastic. The only indication for mitotane is palliative therapy of inoperable adrenocortical carcinoma.
Hormonal Agents, Targeted Drugs, and Other Noncytotoxic Anticancer Drugs
As we continue our discussion of anticancer agents, we will focus on two large groups of drugs: hormonal agents and targeted drugs. The hormonal agents, used primarily for breast cancer and prostate cancer, mimic or suppress the actions of endogenous hormones. The so-called targeted drugs bind with specific molecular targets on cancer cells and thereby suppress tumor growth and promote cell death. Unlike the cytotoxic agents discussed previously, many of which are cell-cycle phase specific, the drugs addressed here lack phase specificity. In addition, many of these drugs lack the serious toxicities associated with cytotoxic agents, including bone marrow suppression, stomatitis, alopecia, and severe nausea and vomiting. Nonetheless, most of these have severe toxicities of their own.
Drugs for Breast Cancer
Principal treatment modalities for breast cancer treatment are surgery, radiation, cytotoxic drugs (chemotherapy), and hormonal drugs. Surgery and radiation are considered primary therapy; chemotherapy and hormonal therapy are used as adjuvants. For a woman with early breast cancer, treatment typically consists of surgery (using total mastectomy or partial mastectomy [lumpectomy]) followed by local radiation. After that, chemotherapy is used to kill cells left behind after surgery and radiation and to kill cells that may have metastasized to other sites. Finally, hormonal agents are taken for several years to reduce recurrence. Increasingly, chemotherapy is used before surgery—so-called neoadjuvant therapy—to shrink large tumors, and thereby permit lumpectomy in women who would otherwise require mastectomy. Drugs for adjuvant therapy are shown in Table 82.5.
TABLE 82.5
Drugs for Adjuvant Therapy of Breast Cancer
| Generic Name | Trade Name | Route | Mechanism | Indications | Major Adverse Effects |
| HORMONAL THERAPIES | |||||
| Antiestrogens | |||||
| Tamoxifen | Nolvadex | PO | Blockade of estrogen receptors | ER-positive breast cancer in premenopausal and postmenopausal women | Increased risk for endometrial cancer and thrombosis Hot flashes, fluid retention, vaginal discharge, nausea, vomiting, and menstrual irregularities |
| Toremifene | Fareston | PO | Blockade of estrogen receptors | ER-positive breast cancer in postmenopausal women only | |
| Fulvestrant | Faslodex | IM | Blockade of estrogen receptors | ER-positive breast cancer in postmenopausal women only | |
| Aromatase Inhibitors | |||||
| Anastrozole | Arimidex | PO | Inhibition of estrogen synthesis | ER-positive breast cancer in postmenopausal women only | Musculoskeletal pain, osteoporosis and related fractures |
| Letrozole | Femara | PO | |||
| Exemestane | Aromasin | PO | |||
| OTHER DRUGS FOR BREAST CANCER | |||||
| Anti-HER2 Antibodies | |||||
| Trastuzumab | Herceptin | IV | Blockade of HER2 receptors | HER2-positive breast cancer in premenopausal and postmenopausal women | Cardiotoxicity and hypersensitivity reactions |
| Ado-trastuzumab | Kadcyla | IV | Blockade of HER2 receptors | HER2-positive breast cancer | Hepatotoxicity, cardiotoxicity, neurotoxicity |
| Pertuzumab | Perjeta | IV | Blockade of HER2 receptors | HER2-positive breast cancer | Cardiotoxicity, hypersensitivity reactions |
| Kinase Inhibitor | |||||
| Lapatinib | Tykerb | PO | Inhibits HER2 tyrosine kinase and EGFR tyrosine kinase | HER2-positive breast cancer in premenopausal and postmenopausal women | Diarrhea, hepatotoxicity, cardiotoxicity, interstitial lung disease |
| Cytotoxic Drugs (Representative Agents) | |||||
| Doxorubicin plus cyclophosphamide | Adriamycin; Cytoxan, Neosar | IV | Direct cell kill by DNA intercalation, topoisomerase II inhibition, and DNA alkylation | Breast cancer in all women, regardless of ER, HER2, or menopausal status | Together, these drugs can cause cardiotoxicity, bone marrow suppression, alopecia, oral and GI ulceration, and hemorrhagic cystitis |
| Paclitaxel | Taxol  , Onxol, Abraxane , Onxol, Abraxane | IV | Direct cell kill by mitotic arrest | Breast cancer in all women, regardless of ER, HER2, or menopausal status | Bone marrow suppression, peripheral neuropathy, alopecia, cardiotoxicity, muscle and joint pain Severe hypersensitivity reactions with Taxol |
| Eribulin | Halaven | IV | Direct cell kill by mitotic arrest | Breast cancer in all women, regardless of ER, HER2, or menopausal status | Bone marrow suppression, peripheral neuropathy |
| Drugs to Delay Skeletal Events | |||||
| Zoledronate | Zometa* | IV | Inhibits osteoclast function | Hypercalcemia of malignancy, prevention of malignancy-related skeletal events | Kidney damage, osteonecrosis of the jaw, rare atrial fibrillation |
| Denosumab | Xgeva† | SubQ | Inhibits osteoclast function and production | Hypercalcemia of malignancy, prevention of malignancy-related skeletal events | Hypocalcemia, serious infections, skin reactions, osteonecrosis of the jaw |
 and Onxol, but not Abraxane
and Onxol, but not AbraxaneHormonal agents for breast cancer fall into two major groups: antiestrogens (e.g., tamoxifen [Nolvadex]) and aromatase inhibitors (e.g., anastrozole [Arimidex]). Antiestrogens block receptors for estrogen, whereas aromatase inhibitors block estrogen biosynthesis. In both cases, tumor cells are deprived of the estrogen they need for growth. However, there is a caveat: for these drugs to work, tumor cells must have estrogen receptors (ERs). Fortunately, most breast cancers are ER positive.
In addition to chemotherapy and hormonal therapy, four other drugs—trastuzumab [Herceptin], ado-trastuzumab emtansine [Kadcyla], pertuzumab [Perjeta], and lapatinib [Tykerb]—can be used for adjuvant treatment. Trastuzumab, pertuzumab, and ado-trastuzumab emtansine block receptors known as human epidermal growth factor receptor type 2 (HER2). In addition, when ado-trastuzumab emtansine binds with HER2 receptors, it releases cytotoxic catabolites that cause cell apoptosis. Lapatinib inhibits two enzymes, known as HER2 tyrosine kinase and epidermal growth factor receptor (EGFR) tyrosine kinase. These drugs are indicated only for cancers that are HER2 positive. Lastly, patients may take denosumab [Xgeva] or zoledronate [Zometa] to minimize hypercalcemia (caused by bone metastases) and fractures (caused by bone metastases as well as hormonal therapy).
What about breast cancer prevention? Currently, two drugs are approved for preventing breast cancer in women at high risk. Both drugs are selective estrogen receptor modulators, or SERMS. One of the drugs—raloxifene [Evista]—is approved only for postmenopausal women. The other drug—tamoxifen [Nolvadex]—is approved for premenopausal and postmenopausal women. Raloxifene is discussed in Chapter 59. Tamoxifen is discussed later in this chapter. Another drug—exemestane [Aromasin] (discussed later)—can also prevent breast cancer, but currently it is only approved for breast cancer treatment.
Antiestrogens
Antiestrogens are drugs that block ERs and hence only work against cells that are ER positive. Benefits derive from depriving tumor cells of the growth-promoting influence of estrogen. Three antiestrogens—tamoxifen, toremifene, and fulvestrant—are approved for adjuvant treatment. Of these, tamoxifen is by far the most widely used.
Tamoxifen
Tamoxifen [Nolvadex] is considered the gold standard for endocrine treatment of breast cancer. The drug is approved for treating established disease and for primary prevention in women at high risk. As discussed later, tamoxifen is a prodrug that must be converted to active metabolites.
Overview of Actions
Tamoxifen blocks ERs in some tissues and activates them in others. Receptor blockade underlies benefits in breast cancer and also underlies some adverse effects (especially hot flashes). Receptor activation leads to other beneficial effects (increased bone mineral density, reduction of low-density lipoprotein cholesterol, elevation of high-density lipoprotein cholesterol) as well as certain adverse effects (endometrial cancer and blood clots). Because tamoxifen can cause receptor activation as well as blockade, the drug is often classified as a SERM.
Mechanism of Action in Breast Cancer
Tamoxifen is a prodrug that undergoes hepatic conversion to active metabolites. These metabolites then block ERs on breast cancer cells and thereby prevent receptor activation by estradiol, the principal endogenous estrogen. Estrogen acts on tumor cells to stimulate growth and proliferation. Hence, in the absence of the influence of estradiol, the rate of tumor cell proliferation declines. Tumors regress in size as the rate of cell death outpaces new cell production. Obviously, if treatment is to be effective, target cells must be ER positive.
Use for Treatment of Breast Cancer
Tamoxifen has two treatment applications: (1) as adjuvant therapy to suppress growth of residual cancer cells after surgery and (2) as treatment of metastatic disease. Tamoxifen can be used in both premenopausal and postmenopausal women.
Use for Prevention of Breast Cancer
Tamoxifen is approved for reducing the development of breast cancer in healthy women at high risk. Unfortunately, tamoxifen increases the incidence of endometrial cancer, pulmonary embolism, and deep vein thrombosis. Hence women considering tamoxifen for chemoprevention must carefully weigh the benefits of treatment (reduced risk for breast cancer) against the risks (increased risk for endometrial cancer and thromboembolic events). According to guidelines issued in 2013 by the U.S. Preventive Services Task Force (USPSTF), tamoxifen chemoprevention is appropriate only for women at high risk, and not for women at low to moderate risk.
To help determine who is at high risk for breast cancer, the National Cancer Institute has created an Internet-based Breast Cancer Risk Assessment Tool. You can access the tool at www.cancer.gov/bcrisktool.
Pharmacokinetics
Tamoxifen is readily absorbed after oral administration. In the liver, CYP2D6 (the 2D6 isoenzyme of cytochrome P450) converts tamoxifen to two active metabolites: 4-hydroxy-N-desmethyltamoxifen (endoxifen) and 4-hydroxytamoxifen. The half-lives of tamoxifen and its metabolites range from 1 to 2 weeks. Because clearance is slow, once-daily dosing is adequate. When treatment is stopped, tamoxifen and its metabolites can be detected in serum for weeks.
Not surprisingly, benefits of tamoxifen are greatly reduced in women with an inherited deficiency in the gene that codes for CYP2D6. Cancer recurrence rate in poor metabolizers may be higher than in good metabolizers. Between 8% and 10% of white women have gene variants that prevent them from converting tamoxifen to its active metabolites. However, at this time, the U.S. Food and Drug Administration (FDA) neither requires nor recommends testing for variants in the CYP2D6 gene, although a test kit is available.
Adverse Effects
The most common adverse effects are hot flashes, fluid retention, vaginal discharge, nausea, vomiting, and menstrual irregularities. In women with bone metastases, tamoxifen may cause transient hypercalcemia and a flare in bone pain. Because of its estrogen agonist actions, tamoxifen poses a small risk for thromboembolic events, including deep vein thrombosis, pulmonary embolism, and stroke.
Perhaps the biggest concern is endometrial cancer. Tamoxifen acts as an estrogen agonist at receptors in the uterus, causing proliferation of endometrial tissue. Proliferation initially results in endometrial hyperplasia and may eventually lead to endometrial cancer. In women taking tamoxifen to treat breast cancer, the benefits clearly outweigh this risk. However, in women taking the drug to prevent breast cancer, the risk/benefit balance is less obvious. In postmenopausal women, endometrial cancer is usually caught early, owing to abnormal menstrual bleeding.
Tamoxifen can harm the developing fetus and hence is classified in FDA Pregnancy Risk Category D. Accordingly, women using the drug should avoid getting pregnant.
Interaction With CYP2D6 Inhibitors
Inhibitors of CYP2D6 can prevent activation of tamoxifen and can thereby negate the benefits of treatment. Put another way, when tamoxifen is combined with a CYP2D6 inhibitor, the risk for breast cancer recurrence is greater than when tamoxifen is used alone. Accordingly, women using tamoxifen should avoid strong CYP2D6 inhibitors. Important among these are fluoxetine [Prozac], paroxetine [Paxil, Pexeva], and sertraline [Zoloft]—selective serotonin reuptake inhibitors (SSRIs) taken by many women to suppress tamoxifen-induced hot flashes. Fortunately, alternatives with less effect on CYP2D6 are available. Among these are escitalopram [Lexapro, Cipralex] (an SSRI) and venlafaxine [Effexor] (a serotonin/norepinephrine reuptake inhibitor).
Toremifene
Actions and Use
Toremifene [Fareston] is an antiestrogen indicated for metastatic breast cancer in postmenopausal women with ER-positive tumors or tumors for which ER status is unknown. The drug is a structural analog of tamoxifen and shares most of that drug’s properties. Like tamoxifen, toremifene is a SERM with antiestrogenic actions in some tissues and estrogenic actions in others. In women with breast cancer, toremifene blocks ERs on tumor cells, thereby depriving them of estrogen’s growth-promoting effects.
Pharmacokinetics
Toremifene is well absorbed after oral administration. Plasma levels peak in 3 hours. The drug undergoes extensive hepatic metabolism, primarily by CYP3A4 (the 3A4 isoenzyme of cytochrome P450). Metabolites are excreted in the feces. The half-life is prolonged (about 5 days), owing to enterohepatic recirculation. As with tamoxifen, drugs that induce CYP3A4 will reduce toremifene levels, and drugs that inhibit the enzyme will raise toremifene levels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


