Chapter Thirty-Three. Anaemia and clotting disorders
CHAPTER CONTENTS
Anaemia
Worldwide, the effects of anaemia and clotting disorders on maternal and fetal morbidity and mortality are enormous. This chapter examines these two clinical problems in detail. Anaemia is reduction in the oxygen-carrying capacity of the blood, which may be due to a reduced number of red blood cells, a low concentration of haemoglobin (Hb) or a combination of both (Bewley 2004). The effects of anaemia involve both mother and fetus. The mother may develop symptoms such as dyspnoea, fainting fatigue, tachycardia and palpitations. She may have reduced resistance to infection and her life may be threatened by antepartum or postpartum haemorrhage.
The fetus may suffer intrauterine hypoxia and growth restriction although it is difficult to separate the effects of anaemia from other factors such as social class, smoking and maternal age. Godfrey et al (1991) found that large placental weight with a reduction in fetal weight was associated with iron-deficiency anaemia. This correlated the change in placental/fetal weight ratio with a risk of hypertension in later life (Godfrey & Barker 1995).
Recognition and incidence of anaemia
The World Health Organization (WHO 2008) set criteria for diagnosis of anaemia in pregnancy as a haemoglobin (Hb) of <11 g/dl, but because of increased understanding of the physiological changes in pregnancy many doctors only investigate women with an Hb of ≤10.0 g/dl (Bewley 2004). The incidence is high in developing countries and is associated with helminth infections, poor nutrition and malaria (Walraven 2008). The WHO state that the prevalence of anaemia in pregnancy in developing countries is between 35% and 75%; however, in developed countries this is lower, at 18%.
Types of anaemia include:
• Iron-deficiency anaemia (IDA).
• Folic acid deficiency.
• Hereditary haemoglobinopathies, sickle cell anaemias and the thalassaemias.
• Anaemia due to blood loss.
Iron-deficiency anaemia
Pathology
Iron is essential for the bioavailability of oxygen to cells. Diminished iron levels are due to poor intake of available dietary iron (Conrad 2008). It is absorbed by the small intestine more readily in pregnancy (Milman 2006). Iron-deficiency anaemia (IDA) is a common pathology of pregnancy, but may be asymptomatic and difficult to diagnose. The physiological changes of blood plasma volume expansion make it appear that haemoglobin is lower. In pregnancy there is a greater demand for iron for haemoglobin synthesis, and demands increase to 7.5 mg/day by the end of pregnancy (Milman 2006). If haemoglobin is low, there is a poor red cell uptake of oxygen and poor oxygen delivery to the placental bed and fetus. The fetus obtains its iron from transferrin in the maternal blood across the placental–maternal interface, usually after 30 weeks of pregnancy. In the earlier weeks maternal iron consumption increases and should meet the later demands, but if iron stores as ferritin are low the demand may not be met. To ascertain those women at risk of IDA, the midwife’s booking interview should highlight the following (Coggins 2001, Conrad 2008):
• Reduced food intake or malabsorption of iron or protein.
• Blood loss from previous heavy menses.
• Iron deprivation from previous pregnancies or short pregnancy gap.
• Multiple pregnancy.
• Chronic urinary tract infection (low iron status affects immunity).
• Previous antepartum or postpartum haemorrhage.
• Women from low social groups.
In a multicultural society such as Britain the likelihood of the inherited haemoglobinopathies should be considered as well as the effects of nutrition and infection on iron status.
Investigations
Identification of IDA involves screening, blood counts, history taking and investigations. Screening all pregnant women for haemoglobin (Hb) concentration regularly indicates the presence of anaemia but will not identify the cause. However, normal non-pregnant reference values may not consider the haemodilution of pregnancy and there may be a danger of overdiagnosing asymptomatic women with low Hb. Women who are identified as having a low Hb should be questioned about nutritional habits, gastrointestinal upset, and excessive menstrual bleeding prior to pregnancy.
IDA is a microcytic anaemia with a fall in mean cell volume (MCV), and a fall in serum ferritin (iron stores) before a fall in Hb. A fall in Hb is a late sign when iron stores have already been depleted. A urine sample should be obtained to exclude urinary tract infection as ferritin levels may be artificially high when an infection is present (Breymann 2005). Coggins (2001) outlined the parameters considered when making a diagnosis of IDA (Table 33.1).
| Blood test | Normal reference range | Validity in diagnosis |
|---|---|---|
| Haemoglobin | 11–15 g/dl (pregnant) | Lacks specificity, affected by haemodilution and smoking |
| Mean cell volume (MCV) | 75–99 fl (femtolitres) | Raised in pregnancy, decreased in IDA |
| Reticulocyte count | 25–75 × 10 9/L | Increased by pregnancy, decreased by IDA |
| Serum ferritin | 15–300 μg/L | Signifies iron stores, early indication of iron deficiency |
| Total iron-binding capacity | 45–72 μmol/L | Non-specific in pregnancy, raised by pregnancy, false positive if infection present |
| Serum iron | 13–27 μmol/L | Decreased by pregnancy, diurnal rhythm, non-specific in pregnancy |
Management
An oral iron preparation is usually given once IDA has been diagnosed. Absorption is maximised if taken with orange juice (Coggins 2001). The daily amount of iron needed to treat IDA is 60–120 mg in divided doses. Oral ferrous salts are more absorbable than ferric salts but all iron preparations tend to have side-effects such as nausea, vomiting and constipation, although in a randomised double blind study of 404 women in four groups it was found that this was not true (Milman et al 2006). In most units in the UK, women are only given iron preparations if IDA has been diagnosed. Iron can be given by intramuscular injection or intravenous infusion if necessary (BNF 2007):
• Intramuscular iron is given in the form of iron sorbitol 50 mg/ml. The dose is 1.5 mg/kg body weight and it can be given daily or weekly. A deep intramuscular injection should be given to avoid staining the skin and fat necrosis.
• Intravenous iron is given as a total dose iron infusion in the form of iron dextran 50 mg/ml. It is given slowly in normal saline and the dose depends on body weight and the degree of iron deficiency. Anaphylactic shock is a major side-effect and intravenous iron should only be given if absolutely necessary.
The treatment for IDA has been reviewed by Cuervo & Mahomed (2003) who looked at 53 trials. Evidence about the effects of iron therapy in pregnancy was inconclusive as there was a shortage of quality trials. Intravenous iron was associated with an increased risk of venous thrombosis. The debate continues as to whether or not to routinely supplement with iron. As the babies of iron-depleted mothers may be adversely affected by their mother’s low iron status supplementation would perhaps be beneficial (Müngen 2003).
Folic acid deficiency anaemia
Incidence
Folic acid is necessary for red cell proliferation and DNA synthesis, and in pregnancy demands are high as the fetus develops and grows (Blackburn 2007). Deficiency may occur in pregnancy in the undernourished, in multiple pregnancy, those on anticoagulants or anticonvulsants, heavy drinkers or smokers causing a megaloblastic anaemia (Bewley 2004). Positive diagnosis cannot be made without a bone marrow biopsy. Demands for folate (naturally occurring) and folic acid (synthesised) are high in pregnancy and many women will have low reserves to call on as the fetus grows (Tsunenobu & Picciano 2006). Synthesised folic acid is more readily acquired by the body systems than folate; hence supplementation may be required and is more successful in raising levels in pregnancy.
Investigations
The red cells are macrocytic (larger) and may be misshapen, fewer in number and the Hb level is low. Plasma folate and red cell folate can be estimated. There may be a low platelet count and white cell count. Serum folic acid is lower than 4 μg/ml (Bewley 2004).
Management
The anaemia usually responds to folic acid supplementation of 5–15 mg folic acid daily. Prevention of anaemia by administration of prophylactic folic acid 300–500 μg daily can be given to those:
• With malabsorption syndrome.
• With haemoglobinopathies (see below).
• Who are on anticonvulsant therapy.
• With a multiple pregnancy.
Folate deficiency is involved in the causation of neural tube defects (Box 33.1).
 33.1
33.1 NEURAL TUBE DEFECTS IN THE FETUS AND FOLATE DEFICIENCY
Neural tube defects (NTDs) include conditions such as anencephaly and spina bifida with or without a meningocele, most of which occur from failure of closure of the caudal neuropore between 22 and 30 days of pregnancy. This leaves the spinal cord unprotected by the spinal column. α-Fetoprotein (AFP) is a fetal protein found in small amounts in maternal serum in normal pregnancies. Any open fetal defect leads to the leaking of AFP into the amniotic fluid, with higher levels than usual entering maternal serum. Higher levels of AFP occur if the gestational age is over-assessed or if more than one fetus is present.
The routine use of ultrasound at booking, then a further anomaly scan, identifies most of these defects so the parents can make a choice between continuing the pregnancy or termination (Devane & Devane 2000). The cause may involve both genetic and environmental triggers. A dietary factor was suspected for a long time and folic acid was implicated as early as 1964 (Wald 1991).
The Medical Research Council (MRC) Vitamin Study Group (1991) undertook a multicentred, double-blind, randomised trial across 33 centres in seven countries to see if supplementation with folic acid or a mixture of seven other vitamins around the time of conception could prevent NTDs. The findings were that folic acid supplements prevented three-quarters of the cases of NTD recurrence.
Folic acid is used in the metabolic chain to provide the chemical bases of three of the essential DNA components: guanine, adenine and thymine. Vitamin B 12 is necessary to form an enzyme in the metabolic pathway of folate. Women who are epileptic tend to have more congenital abnormalities, in part due to the antiepileptic medication altering the absorption of folate (Steegers-Theunissen 1995).
Following the findings of the above study the Department of Health (1992) recommended the use of 4 mg of folic acid 3 months prior to conception continued until the early months of pregnancy. However, those women at risk, under-nourished, of low economic status and those with unplanned pregnancies would be unlikely to supplement diet with folic acid (Eichholzer 2006). Some foods such as cereals are fortified in the UK; however, fortification in USA and Canada of flour and cereals since 1998 has proven the benefits with no evidence of risk. Despite the RCOG recommendations to fortify bread flour in the UK this has not yet been agreed (Food Standards Agency 2008, RCOG 2008).
Haemoglobinopathies
Two of the most common diseases are the recessively inherited sickle cell disease and thalassaemias which affect haemoglobin synthesis. In utero, the fetus is not affected as it carries fetal haemoglobin (HbF) which has a greater affinity to oxygen, but soon after birth the switch between fetal and adult haemoglobin (HbA) begins (Bailey & Gwinnutt 2008). As the ratio of HbF to HbA changes, the symptoms of inherited disease become evident (Stephen & Cunningham 1998).
The globin chains
The gene for the α-globin chain family is located on chromosome 16 and for the β-globin chain family on chromosome 11. The α-globin chain is 141 amino acids long and the β-globin chain is 146 amino acids long. All haemoglobin variants have a tetrameric structure with four protein chains in association with four haem molecules. The four protein chains in normal haemoglobin take up a particular shape which allows maximum uptake, delivery and release of oxygen into the tissues.
Inherited genes produce abnormal proteins that cannot carry out their function efficiently resulting in ill-health with anaemia, hypoxia, tissue damage and haemolysis (Fig. 33.1). There are three forms of inherited haemoglobinopathies:
• Structural Hb variants, in which there is a fault in either the α-globin chain or the β-globin chain.
• The thalassaemias, in which there is reduced production of either the α-globin chain or the β-globin chain.
• Failure to switch from the production of HbF to HbA, which is clinically insignificant, although in some instances helps the sufferer because oxygen is absorbed more easily by HbF (Weatherall 1997b).
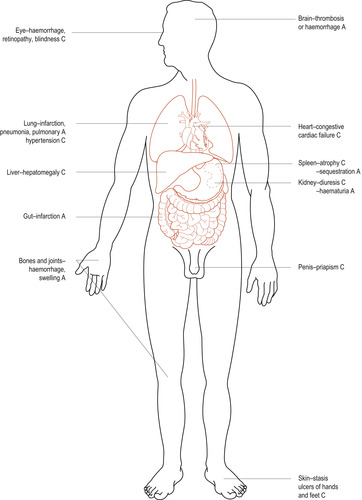 |
| Figure 33.1 Major clinical manifestations of sickle cell anaemia. A, acute; C, chronic. |
Sickle cell disease
Sickle cell disease (SCD) is the most common of the structural haemoglobin defects (Weatherall 1997a) (Table 33.2). A single amino acid substitution of valine for glutamic acid results in a haemoglobin molecule that is less soluble. When oxygen is low, the molecules form long, linear stacks that distort the red cells into a sickle shape. The inheritance of one gene from each parent (homozygous genotype for HbSS) makes the individual sickle cell-positive. Those who inherit one gene (heterozygote) have the sickle cell trait (HbAS) and usually do not display signs of the disease. They do, however, have some protection against the organism Plasmodium falciparum, the cause of a severe form of malaria. The malarial parasite enters the red blood cell and makes it sickle. This cell and its parasite are destroyed by the spleen, protecting the individual from malaria (Frenette & Atweh 2007).
| Haemoglobin | Disease |
|---|---|
| HbSS | Homozygous sickle cell disease (sickle cell anaemia) |
| HbSC | Heterozygous sickle cell disease (sickle cell C disease), mild anaemia, and fewer crises, risk of retinal damage and thromboembolic problems in pregnancy |
| HbCC | Homozygous CC disease (not a sickling disorder) |
| HbS β-thalassaemia | Sickle β-thalassaemia, generally produces sickle Hb |
| HbAS | Sickle cell trait, generally no problems |
Heterozygote parents have a 50% chance of producing an infant affected by SCD (Sickle Cell Society 2008). Diagnosis may be made in the first trimester of pregnancy by chorionic villus sampling. Molecular technology in the future may be able to diagnose SCD with a few fetal cells from maternal blood (Frenette & Atweh 2007).
Incidence
The incidence of SCD varies from country to country and is about 1 in 4 in parts of Africa and 1 in 10 of the black population of the USA and UK. It is estimated that 50 000 Americans have SCD. Amongst African-Americans 1:375 have HbSS, 1:832 have HbSC and 1:1667 have Hb β-thalassaemia (Sickle Cell Society 2008). Heterozygote incidence ranges from 0.9% in Europe to 13.3% in Africa. The incidence of SCD in the UK is 1 in 2400 live births and is the ‘fastest growing genetic disorder’ (Sickle Cell Society 2008). Other variants of the disease are being found and crossing racial and ethnic groups so it is no longer appropriate to assume that a particular group will have SCD. Routine screening is already taking place both antenatally and in the newborn (Anglin 2007a, Zack-Williams 2007).
Pathophysiology
Deoxygenation is the most common cause of sickling; HbSS reacts by creating non-pliable intracellular fibres which pull the cells into a banana shape or holly leaf shape. These block capillaries, creating the pain of a sickle cell crisis (Bailey & Gwinnutt 2008). Decreased plasma volume, hypothermia, infection and acidosis also precipitate sickling. This will occur with minor degrees of oxygen shortage in people with sickle cell disease, but lack of oxygen has to be severe to cause sickling in people with sickle cell trait.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


