- The aminoglycoside antibiotics are potent bactericidal agents that are rapidly lethal to bacteria in a concentration-dependent manner; their therapeutic potential for widespread use is restricted by ototoxicity and nephrotoxicity.
- Aminoglycosides have a wide spectrum of activity and are used for the treatment of many Gram negative and Gram positive bacterial infections; they are sometimes used in combination with β-lactam antibiotics.
- For further information, see Vakulenko and Mobashery (2003).
Aminoglycoside antibiotics used clinically include gentamicin, neomycin, streptomycin, tobramycin, and amikacin; their structures are shown in Figure 4.1.1 and their therapeutic indications are listed in Table 4.1.1.
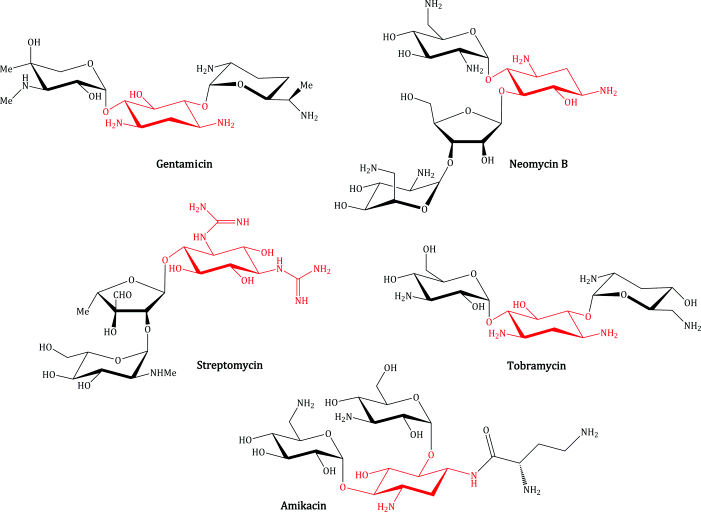
Table 4.1.1 Therapeutic indications for the aminoglycoside antibiotics.
| Aminoglycoside antibiotic | Indications |
| Gentamicin | Septicaemia, burns, endocarditis, used topically to treat eye and ear infections, pneumonia in hospital patients, Listeria meningitis |
| Neomycin | Suppression of intestinal bacteria prior to colonic surgery |
| Streptomycin | M. tuberculosis |
| Tobramycin | Chronic pulmonary P. aeruginosa infection in cystic fibrosis |
| Amikacin | Serious infections caused by Gram negative bacteria resistant to gentamicin |
4.1.1 Discovery
The first aminoglycoside antibiotic was discovered as the result of a systematic study of soil microbes by Selman Waksman and his co-workers, who were searching for an agent with activity against tuberculosis (TB). It had previously been noticed that certain microorganisms predominated in soil and it was suggested that these species must be producing an antibiotic agent in order to afford them survival superiority. Waksman and co-workers carried out over 10 000 careful culture experiments before they discovered the aminoglycoside agent, streptomycin, which is produced by an actinomycete mould, Streptomyces griseus, and is active against the TB-causing bacterium Mycobacterium tuberculosis (Schatz and Waksman, 1944; Schatz et al., 1944). Streptomycin was first used clinically in 1944, when its use led to the curing of a young woman with pulmonary TB; at a time when TB was essentially a slow death sentence, this cure was astonishing.
Waksman was awarded the Nobel Prize in Physiology or Medicine in 1952 ‘for his discovery of streptomycin, the first antibiotic effective against tuberculosis.’ Sadly, this award was the subject of some controversy, as Albert Schatz, who had carried out the experiments that resulted in the discovery of streptomycin while working as a PhD student under Waksman’s guidance, argued that the Nobel Prize should also have been awarded to him for his part in its discovery (Kingston, 2000, 2004; Ainsworth, 2006).
The search for new antibiotics for the treatment of bacterial infections, particularly TB, was continued by Waksman and his co-workers and other international groups. The Streptomyces family proved to be a rich source of antibiotics, resulting in the discovery of antibacterial agents belonging to several structural classes. Besides streptomycin, other key aminoglycoside discoveries were: neomycin from S. fradiae in 1949 (Waksman and Lechevalier, 1949; Waksman et al., 1949); paromomycin from Streptomyces rimosus in 1959 (Coffey et al., 1959); kanamycin from Streptomyces kanamyceticus in 1957 (Maeda et al., 1957; Takeuchi et al., 1957; Umezawa et al., 1957); and tobramycin – initially from Streptomyces tenebrarius in 1968 (Stark et al., 1968), but which can also be obtained through the synthetic modification of kanamycin B (Takagi et al., 1973, 1976).
Another aminoglycoside, gentamicin, was isolated in 1963 (Weinstein et al., 1963) from various species of Micromonospora bacteria, as a complex of several structurally related agents. There are three forms, of which the most often encountered clinically is gentamicin C, consisting of gentamicins C1, C1a, C2, and C2b (Wagman et al., 1967). The discovery of sisomicin, isolated from Micromonospora inyoensis in 1970, added to the aminoglycoside arsenal (Weinstein et al., 1970), while further aminoglycosides have been made by synthetic modifications of the original natural aminoglycosides (more about this later). Of these, amikacin was found to have improved pharmacokinetic and pharmacodynamic parameters (Gooding et al., 1976), in addition to excellent clinical efficacy and a lower incidence of bacterial resistance, and so was adopted for clinical use.
You may be wondering why some aminoglycosides end in –mycin and others with –micin. Well, it relates to the microorganism which is the source of each agent; those from a Streptomyces species are given the ending –mycin, while those agents from a Micromonospora species have –micin at the end of their name. While this is a useful guide to the origin of an aminoglycoside, it does not necessarily extend to the naming of other microorganism-derived agents and there are many examples of non-aminoglycoside agents from Streptomyces species that are not named by the same system, such as chloramphenicol, novobiocin, cycloserine, and clavulanic acid.
Structurally, the antibiotic aminoglycosides are glycosidic polycyclic structures, incorporating an aminocyclitol ring (streptamine, streptidine, or 2-deoxystreptamine) (Figure 4.1.2), of which the latter is most commonly found in clinical aminoglycosides, usually attached to two or three aminosugar rings. If you look back at Figure 4.1.1, you can see the aglycone1 aminocyclitol ring in each structure – it is easily identifiable as it does not have an O atom as part of the ring (see Figure 4.1.2); the other cyclic parts of each molecule are sugars, recognisable as the five- or six-membered cyclic systems that include an O atom in the ring.
Figure 4.1.2 The aminocyclitol ring systems found in the aminoglycoside antibiotics, 2-deoxystreptamine being the most common in clinical aminoglycosides
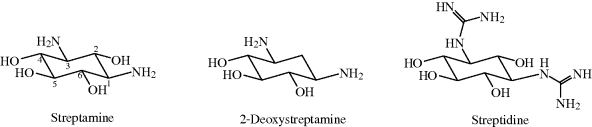
The aminoglycosides can be categorised into related groups based upon the glycosidic linkages of the cyclic aminosugar components to the central aminocyclitol ring: the 4,5-disubstituted group includes neomycin and paromomycin; the 4,6-disubstituted group includes the kanamycins, gentamicins and tobramycin; while the original aminoglycoside, streptomycin, has a six-membered monosubstituted aminocyclitol ring.
The possibility that the genetic sequencing of many bacteria may lead to the discovery of further aminoglycosides (and other classes of antibiotics) has been recognised, but no new agents have resulted from such genetic studies thus far (Paradkar et al., 2003; Kudo and Eguchi, 2009).
Have another look at the structures in Figure 4.1.1 and identify how many ionisable groups each aminoglycoside possesses. How many do you think will be ionised and what will be the overall charge on each molecule at pH 7.0? We will come back to these questions in Subsections 4.1.3 and 4.1.4, when we consider the bioavailability and the mode of action of these agents, which are both linked to ionisation.
4.1.2 Synthesis
In addition to ionisable groups, how many stereogenic, or chiral, centres can you find in each structure in Figure 4.1.1?2 Having many chiral centres, each aminoglycoside represents a significant synthetic challenge. As these compounds were originally isolated from microorganisms, a biotechnological approach to their production, similar to that employed in the production of other particularly complex antibiotics (for examples, see Subsections 2.2.2 and 5.1.2), is the preferred method. In fact, Waksman and Schatz patented their original method for the production of streptomycin and licensed it to Merck (Waksman and Schatz, 1948). Many other patents followed, for example those from Pfizer (Ratajak and Nubel, 1959) and Ayerst (now Wyeth) (Trussel, 1951). Fermentation conditions have been well studied and remain the subject of research aimed at optimising the yields and purity of the aminoglycosides produced (Sánchez et al., 2010). Full knowledge of the biosynthetic pathways would aid optimal fermentation, but they remain unclear in parts, despite considerable study (Llewellyn and Spencer, 2006; Flatt and Mahmud, 2007). The semi-synthetic aminoglycosides, such as amikacin, are produced commercially by chemical modification of the natural precursor, in this case kanamycin A, which is obtained from fermentation (Mangia, 1998).
As we have previously seen, some chemists view a complex chemical structure, such as those of the aminoglycosides, as a synthetic challenge to be overcome. You will probably not be surprised then to learn that the total synthesis of several members of the aminoglycoside family has been accomplished, although never in sufficient quantities to represent a viable production route. You’ve probably already realised, by considering the structures of the aminoglycosides in Figure 4.1.1, that with so many chiral centres and amino and hydroxyl groups, careful synthetic and protection strategies were required in these total syntheses. If those challenges were not enough, to make it even more difficult, each aminoglycoside consists of a different arrangement of substituents around the aminocyclitol ring, different glycosidic components attached to the aminocyclitol ring, and different substituents, with various relative substitution sites and stereochemistries, on the aminosugar rings. It is rather impressive that structures of such complexity were synthesised in the late 1960s and 1970s, when many of the elegant synthetic methods now available had not even been thought of. Much of the total synthesis of the aminoglycosides was carried out by Umezawa and co-workers and resulted in a number of synthetic routes that allowed the synthesis of the main clinical aminoglycosides and the evaluation of their essential features (Umezawa, 1974, 1978, 1979).
To understand fully the difficulty of these total syntheses, we have to consider some of the problems faced by carbohydrate chemists, which are varied and considerable in number and complexity. Any simple carbohydrate, such as a monosaccharide like glucose, has several hydroxyl groups that must be differentiated in order to react specifically at any particular position; this is normally achieved by a selective protection strategy, in which the relatively close spatial arrangement of particular hydroxyl groups is exploited to protect those groups, leaving others as the free OH groups. The example below will help to illuminate the problems that must be overcome, but for simplicity and brevity, a significant amount of detail that can be found in the original reviews is omitted (Umezawa, 1974, 1978, 1979).
Figure 4.1.3 highlights the component parts of streptomycin. The glycosidic part of streptomycin, streptose, linked to L-glucosamine, is attached at the C4-OH group of the aminocyclitol ring of streptidine. When starting from streptamine, how can this linkage be made specifically at that position without attaching the streptose portion at the C2-OH, C5-OH, or C6-OH positions?
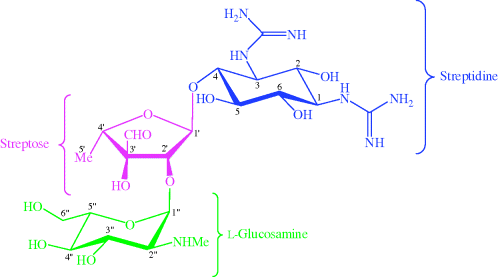
Note that the carbons numbered with a ‘prime’ in Figure 4.1.3 (with ring atom numbers 1′, 2′, 3′, etc.) refer to the sugar (streptose) ring attached to the C4 position of the aminocyclitol systems. It is a general feature of aminoglycosides that the atoms of the ring attached to the C4 position are numbered 1′, 2′, 3′, etc.
The same question may be asked about the linkage between L-glucosamine and streptose: how can it be made specifically between C2′-OH of streptose (as numbered in Figure 4.1.3) and the anomeric position of L-glucosamine (C1″)? For both questions, the answer involves a careful protection strategy to allow only the desired groups to react:
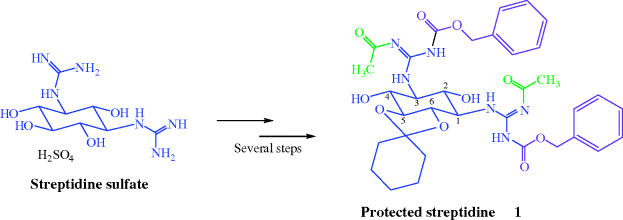
- Addition of acetyl and carbobenzyloxy carbamate (Cbz) protecting groups to each of the guanidine groups, and a cyclic acetal to the C5-OH and C6-OH groups of streptidine, through several steps, produces selectively protected streptidine 1, ready for reaction with the streptose–glucosamine glycosidic unit (Scheme 4.1.1). You will note that protected streptidine 1 has both C2-OH and C4-OH available for reaction when forming the bond to streptose, which could cause a problem and a mixture of products. Although this can and does happen, reaction at C4-OH is preferred, presumably due to steric hindrance at C2-OH caused by the two Cbz groups on the guanidines, so that the major product has the correct substitution at C4-OH and can be readily separated and purified.
- The glycosidic part requires substantially more preparatory work before it can be linked to the protected streptidine 1. To simplify the atom numbering and make it easier to track each atom and its substituents through the synthesis, we will retain the numbering system used in Figure 4.1.3. First, the aldehyde group at C3′ of streptose complicates the protection and synthesis, so the reduced derivative, dihydrostreptose, is used, in which the C3′-CHO group has been reduced to C3′-CH2OH. Dihydrostreptose is selectively protected by benzylation (CH2Ph, Bn) of the anomeric hydroxyl (C1′), which is more reactive than the other hydroxyl groups, then acetalisation of the C3′-OH and C3′-CH2OH groups forms the protected dihydrostreptose derivative 2, with a sole hydroxyl group at C2′ ready for reaction (Scheme 4.1.2).
Scheme 4.1.2 Selective protection of dihydrostreptose and L-glucosamine, followed by glycoside bond formation to obtain the key intermediate dihydrostreptobiosaminide 5 (Umezawa, 1979)
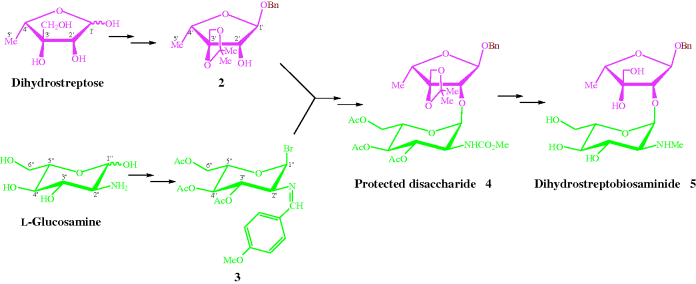
- The protected L-glucosamine 3 can be prepared from L-glucosamine in several steps; again, the anomeric centre is easily and selectively protected first. The amino group at C2″ is protected by Schiff’s base formation, using para-methoxybenzaldehyde, before esterification of the C3″-OH, C4″-OH, and C5″-OH groups. Having protected the other functional groups, the anomeric centre can be deprotected and activated, and the α-bromo derivative 3 can be formed, which is ready for reaction with a nucleophilic hydroxyl group, to form the specific α-glycosidic bond between C2′ of dihydrostreptose and C1″ of glucosamine. Hydrolysis of the Schiff’s base and N-methylation, via a carbamate in protected disaccharide 4, gives the required N1-methylated disaccharide, which is then deprotected, except for the benzyl group on C1′-OH of the dihydrostreptose molecule, to give a protected dihydrostreptobiosaminide 5: a key intermediate (Scheme 4.1.2).
- Several more steps are still required to prepare the disaccharide for coupling to the protected streptamine 1. These reactions follow a sequence of full protection, selective deprotection, and elaboration, before glycosyl chloride 6 is formed, which is reacted with protected streptamine 1, followed by deprotection to dihydrostreptomycin 7. Oxidation of the CH2OH group on C3′ of dihydrostreptose requires yet more protection and deprotection steps before the aldehyde can be reliably formed and streptomycin is finally achieved (Scheme 4.1.3).
Scheme 4.1.3 Final steps in the total synthesis of streptomycin via dihydrostreptomycin 7 (Umezawa, 1979)
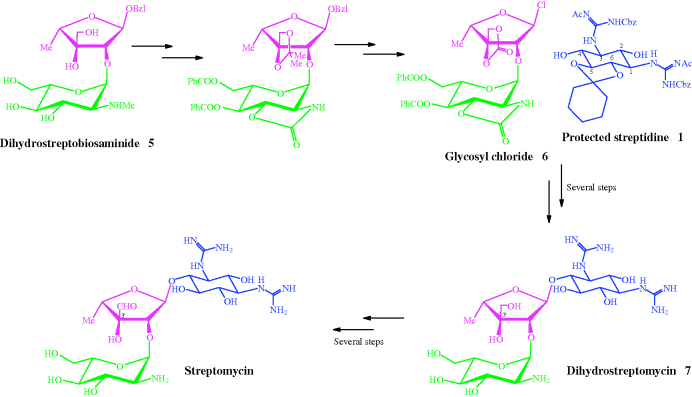
Considering this example was very much abbreviated, you can see that the total synthesis of aminoglycosides is far from a simple task and requires many sequential reactions. As we said earlier, it is thus easier to use the microorganisms that form these species to produce the commercial quantities needed for clinical use.
To investigate the essential structural features for antibacterial activity, selectivity, and efficacy, the synthetic routes outlined above have been further developed to allow the effect of modifications to be examined and pharmacophores to be identified (see, for example, Davies et al., 1978; Igarashi et al., 1982; Hanessian et al., 2010; Yan et al., 2011).
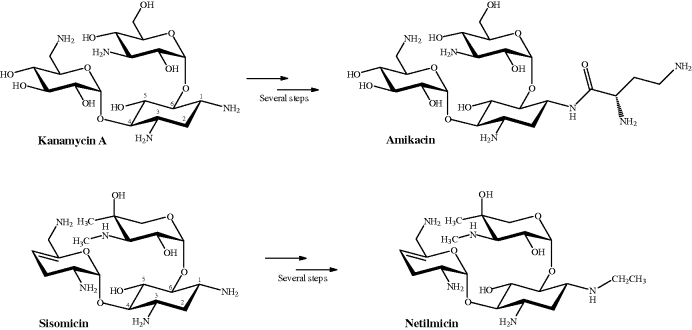
Following the identification of bacterial resistance pathways (more on these later, in Subsection 4.1.5) via conjugation to specific N or O atoms of an aminoglycoside, there has been much research on the modification of the target N and O atoms in order to reduce their susceptibility to metabolism and to avoid resistance; for example, the clinical aminoglycoside agent amikacin is a successful example of synthetic modification to N1 of kanamycin A (Kawaguchi 1976), involving the addition of the 2S-hydroxy-4-aminobutyryl group (achieved by another selective protection strategy (Mangia, 1998; Hanessian et al., 2003)), while netilmicin was synthesised from sisomicin (from the gentamicin family of aminoglycosides) by ethylation of N1 (Scheme 4.1.4) (Wright, 1975; Miller et al., 1976). The latter agents are not currently in clinical use in most countries, due to observed variations in efficacy, toxicity, and pharmacokinetics in different patient groups, and generally greater toxicity than the other aminoglycosides (Price, 1986).
Scheme 4.1.4 Semi-synthetic aminoglycosides: amikacin from kanamycin A and netilmicin from sisomicin
More recently, there has been a considerable amount of research directed at the systematic exploration of structural requirements for activity and for avoiding resistance, particularly when it is due to metabolism (e.g. N-acetylation). The development of synthetic combinatorial methods suitable for the synthesis of many analogous structures has allowed libraries of aminoglycoside derivatives to be created and these methods, the resultant aminoglycosides, and their structure–activity relationships have been investigated (Silva and Carvalho, 2007; Zhou et al., 2007).
4.1.3 Bioavailability
At the end of Subsection 4.1.1, we asked you to consider the structures of the five representative aminoglycosides in Figure 4.1.1 and to decide which were ionisable, which would be ionised at pH 7.0, and what the overall charge on the molecule would be at this pH. You will have found that they all have a number of hydroxyl (OH) and amino (NH2) groups, while streptomycin also has two guanidine groups (-NH-C(=NH)-NH2). Of these, at physiological pH only the amino and guanidino groups are ionisable, each group becoming protonated when the pH is lower than the pKa of that individual amino or guanidino group. Remember that, aliphatic hydroxyl groups (pKa ~ 18–19) are essentially neutral at physiological pH. We would therefore expect the overall charge on each molecule at physiological pH (7.0) to be: gentamicin +5, neomycin B +6, streptomycin +3, tobramycin +5, and amikacin +5, which reflects the number of protonated amino/guanidino groups on each at this pH. In reality, one of the amino groups in each molecule, N3 of the aminocyclitol ring, has a pKa of around 6, such that it is only partly ionised at pH 7, making the overall average charge slightly less than the integer values above (Walter et al., 1999; Jin et al., 2000; Kaul and Pilch, 2002).
When considering their oral bioavailabilities with respect to their state of ionisation, you could predict, from their polycationic character, that the expected absorption of the aminoglycosides after oral administration is likely to be fairly low. In fact, less than 1% of these antibiotics is absorbed across the gastrointestinal (GI) tract following oral administration (Forge and Schacht, 2000), so parenteral routes are commonly used. The poor absorption across the GI tract is very useful, however, in pre-operative bowel cleansing, when these agents are used to limit the risk of post-operative infection due to commensal or opportunistic bacteria.
Systemic aminoglycoside antibacterial treatment is usually achieved through carefully controlled IV infusion, which results in complete absorption and predictable peak serum levels. The latter parameter is particularly important, as infusions that are given too rapidly can result in high peak levels, which increases the risk of ototoxicity, when the cochlear and hair cell binding sites become saturated (Allegaert et al., 2008). Intramuscular injection also results in high bioavailability, but as these very polar molecules do not easily cross membranes, they need to be administered directly to the site of infection. The clinical use of aminoglycosides with respect to their pharmacokinetics has been reviewed recently and recommendations have been made for their optimal use (Avent et al., 2011).
As a result of the structures of these bactericidal agents (Figure 4.1.1), we might expect them to concentrate in the serum and extracellular fluid, with little distribution across membranes into cells and other compartments. In fact, the apparent volume of distribution of aminoglycosides approximates to the extracellular fluid volume, which is about 25% of the total body weight (Duff, 1992).
The use of these agents for the treatment of infections in readily accessible organs, such as their use in the treatment of infective endocarditis (see Subsection 4.1.6.2 below), seems obvious, but how can these agents be used for the treatment of meningitis, which requires therapeutic concentrations to be achieved in the cerebrospinal fluid (CSF)? While the highly polar nature of aminoglycosides suggests that they would not be able to cross the blood–brain barrier, and this is usually the case, this membrane is compromised and more permeable when inflammed in bacterial meningitis. The concentrations of aminoglycosides achieved in the CSF as a result are variable (Lutsar et al., 1998), but appear to be sufficiently high for excellent therapeutic results (Gaillard et al., 1995). Neonates have a more permeable blood–brain barrier and aminoglycoside therapy has been successfully combined with ampicillin for many years (McCracken, 1984). Interestingly, the introduction of a vaccine for Haemophilus influenzae B (Hib), a common cause of meningitis, has changed the epidemiology of this threatening infection and may result in the need for different antibiotic combinations as the infective agents change (Kim, 2009). Intralumbar, intrathecal, and intraventricular administration are alternatives for aminoglycoside treatment of meningitis, resulting in more rapid and higher aminoglycoside levels being achieved (Kaiser and McGee, 1975). New methods to improve the delivery of aminoglycosides are under investigation; one that has promising results is the use of liposomes to enhance uptake and delivery (Schiffelers et al., 2001; Rukholm et al., 2006; Alipour et al., 2009).
The polycationic character of aminoglycosides imparts low serum protein binding, making the majority of the dose available for its therapeutic effect, and leads to efficient urinary excretion via the kidneys, with greater than 90% of an aminoglycoside dose being excreted by this route over 24 hours. For susceptible bacteria, the minimum inhibitory concentration (MIC) for most aminoglycosides lies in the range 0.5–5 μg/mL, which is evidence of their considerable potency, while the serum half-lives (t½) of the aminoglycosides are around 2–3 hours (Turnidge, 2003). Due to their concentration-dependent effect, an important pharmacodynamic parameter for aminoglycoside efficacy is the ratio of Cmax to the MIC, which should be around 10–12 for optimal therapeutic benefit (Filho et al., 2007; Turnidge and Paterson, 2007). When used in this ratio, a large, once-daily dose is effective, despite the short half-life of these agents; this efficiency may be linked to the considerable post-antibiotic effect, which suppresses bacterial growth for over 8 hours after aminoglycoside concentrations fall below the MIC, and so enhances the bactericidal effect of the once-daily dosing (Begg and Barclay, 1995).
We should also consider the uptake of aminoglycosides into bacteria in order to exert their bactericidal action. How can sufficient amounts of a molecule so polar cross the complex bacterial cell wall of a Gram negative bacterium to access the ribosomal RNA? Studies have shown that there are several key aspects:
- First, a small amount of aminoglycoside enters the bacterial cells against a concentration gradient by an energy-requiring process (called energy-dependent phase I, EDP-I) during an essential induction phase. The energy is generated by a respiratory chain located on the membrane, which explains why anaerobic bacteria (with poor electron transport systems) are resistant to aminoglycosides and facultative anaerobes can resist low concentrations.
- The internalised molecules then bind to RNA in a second energy-dependent process (energy-dependent phase II, EDP-II) and cause misreading during protein synthesis, which results in catastrophic changes to the cell wall construction, and a second, rapid-uptake phase of aminoglycoside accumulation, which is lethal to the bacterial cells (Vakulenko and Mobashery, 2003).
4.1.4 Mode of Action and Selectivity
Aminoglycosides result in rapid and complete lethality to cells, which is almost independent of the bacterial load, providing that they achieve the minimum bactericidal levels required. They are most active against aerobic bacteria, particularly Gram negative strains, with particular aminoglycosides displaying distinct bactericidal profiles, including activity against some Gram positive strains (Vakulenko and Mobashery, 2003; Magnet and Blanchard, 2005; Jana and Deb, 2006; Shakil et al., 2008).
The aminoglycoside antibiotics act by binding to a part of the 30S subunit of prokaryotic ribosomes, to a region of the highly conserved 16S ribosomal RNA. Most in clinical use are believed to bind at the A site, which accepts the charged tRNAs during the process of translation and is a vital element of the translation mechanism in bacteria. At this point, if you need a reminder, you can refer back to Subsection 1.1.3.3, where the process of translation, which results in protein synthesis, is outlined in schematic form.
In the normal process of translation of mRNA, the correct (termed ‘cognate’) tRNA is selected through two key interactions. First, the codon on the mRNA has to match with the anticodon on the tRNA, and, second, when the cognate tRNA binds to the A site, subtle favourable conformational changes result and enable important interactions between the codon and anticodon RNA double helices, which discriminate between the correct and incorrect (non-cognate) tRNA. An incorrect tRNA, whether non-cognate or near-cognate (with a similar, but incorrect, codon sequence) cannot initiate the same favourable interactions; in this way, only the cognate tRNA is selected and proof-reading is achieved. In particular, two conserved adenine residues, A1492 and A1493, are known to be involved in the normal decoding process; before translation commences, these two adenine residues can be found stacked in the interior of the RNA double helix (in helix 44). Binding of the cognate tRNA causes a conformational change that ‘flips’ the two adenine residues away from their initial position, resulting in the formation of hydrogen bonds from N1 of the two adenines to the tRNA residues in the first two positions of the codon–anticodon triplet. It is these intermolecular interactions that are thought to be responsible for differentiation between cognate and non-cognate tRNAs, as the latter cannot form the same favourable interactions, so are energetically less beneficial (Vakulenko and Mobashery, 2003).
Aminoglycoside binding to the ribosome is initially encouraged by the favourable interactions possible between the positively charged aminoglycoside and the negatively charged phosphate groups of the RNA double helix; tobramycin has been shown to bind as a trication (Jin et al., 2000), while the neomycins have between three and five protonated groups on binding (Kaul and Pilch, 2002; Kaul et al., 2003). These electrostatic interactions are important thermodynamic contributors to the binding. Each class of aminoglycoside has a structure complementary to slightly different sites on the ribosomal RNA, depending upon the exact nature and position of the substituents, resulting in a subtly altered outcome. For example, the 2-deoxystreptamine-based aminoglycosides, such as gentamicin, neomycin, tobramycin, and amikacin, are believed to bind to the A site and adversely affect the accuracy of translation. Streptomycin also induces poor accuracy, but appears to achieve this result in a slightly different way.
How does the binding of an aminoglycoside to the bacterial ribosome lead to misreading of the mRNA (Chittapragada et al., 2009)?
- It has been shown that the 2-deoxystreptamine aminoglycosides bind in the major groove of double-stranded (ds) RNA (Jin et al., 2000), near to A1492 and A1493 in helix 44 (which borders the A site), resulting in displacement of these adenine residues out of the interior of helix 44 and towards the minor groove (Kaul and Pilch, 2002). Several energetically favourable interactions encourage this conformational change.
- The 2-deoxystreptamine ring and the sugar moiety attached at C4 form highly conserved hydrogen bond interactions between N1 and the carbonyl of uracil, U1495, N3 and guanine, G1494, and between the ring O and C6′-NH2 of the ‘primed ring’3 and adenine, A1408. Two conserved electrostatic interactions are also observed between the ionised N3 and the phosphate groups of A1493 and G1494 (Chittapragada et al., 2009).
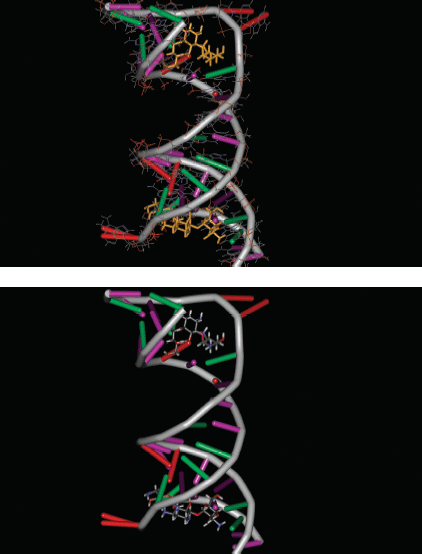
- The new positions of A1492 and A1493 are stabilised by these interactions with the aminoglycoside, which compensates for the less favourable binding of non-cognate tRNA molecules, allowing the incorporation of incorrect amino acids and introducing misreading of the mRNA and reduced proof-reading of the sequence (Vakulenko and Mobashery, 2003; Yang et al., 2006). The binding of tobramycin to a synthetic dsRNA portion of the 30S subunit is illustrated above, where the bases guanine, uracil, cytosine, and adenine can be seen close to the aminoglycoside (Figure 4.1.4). In this model, the binding of tobramycin causes distortion of the dsRNA and mismatching of bases in the sequence used.
Figure 4.1.4 Complex of two molecules of tobramycin with a synthetic putative ribosomal A binding site: (a) dsRNA with tobramycin; (b) with tobramycin colour-coded by atom
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


