Chapter 2
Altered Cellular and Tissue Biology
Kathryn L. McCance, Todd Cameron Grey and George Rodway

Injury to cells and their surrounding environment, called the extracellular matrix, leads to tissue and organ injury. Although the normal cell is restricted by a narrow range of structure and function, it can adapt to physiologic demands or stress to maintain a steady state called homeostasis. Adaptation is a reversible, structural, or functional response to both normal or physiologic conditions and adverse or pathologic conditions. For example, the uterus adapts to pregnancy—a normal physiologic state—by enlarging. Enlargement occurs because of an increase in the size and number of uterine cells. In an adverse condition, such as high blood pressure, myocardial cells are stimulated to enlarge by the increased work of pumping. Like most of the body’s adaptive mechanisms, however, cellular adaptations to adverse conditions are usually only temporarily successful. Severe or long-term stressors overwhelm adaptive processes and cellular injury or death ensues. Altered cellular and tissue biology can result from adaptation, injury, neoplasia, accumulations, aging, or death. (Neoplasia is discussed in Chapters 12, 13, and 14.)
Cellular Adaptation
Cells adapt to their environment to escape and protect themselves from injury. An adapted cell is neither normal nor injured—its condition lies somewhere between these two states. Cellular adaptations, however, are a common and central part of many disease states. In the early stages of a successful adaptive response, cells may have enhanced function; thus it is hard to differentiate a pathologic response from an extreme adaptation to an excessive functional demand. The most significant adaptive changes in cells include atrophy (decrease in cell size), hypertrophy (increase in cell size), hyperplasia (increase in cell number), and metaplasia (reversible replacement of one mature cell type by another less mature cell type). Dysplasia (deranged cellular growth) is not considered a true cellular adaptation but rather an atypical hyperplasia. These changes are shown in Figure 2-1.
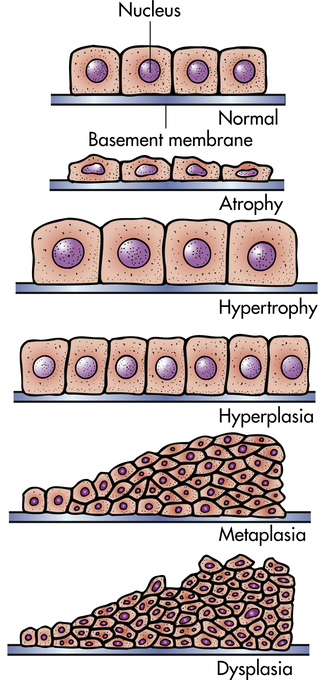
Atrophy
Atrophy is a decrease or shrinkage in cellular size. If atrophy occurs in a sufficient number of an organ’s cells, the entire organ shrinks or becomes atrophic. Atrophy can affect any organ, but it is most common in skeletal muscle, the heart, secondary sex organs, and the brain (Figure 2-2). Atrophy can be classified as physiologic or pathologic. Physiologic atrophy occurs with early development. For example, the thymus gland undergoes physiologic atrophy during childhood. Pathologic atrophy occurs as a result of decreases in workload, use, pressure, blood supply, nutrition, hormonal stimulation, and nervous stimulation. Individuals immobilized in bed for a prolonged time exhibit a type of skeletal muscle atrophy called disuse atrophy. Aging causes brain cells to become atrophic and endocrine-dependent organs, such as the gonads, to shrink as hormonal stimulation decreases. Whether atrophy is caused by normal physiologic conditions or by pathologic conditions, atrophic cells exhibit the same basic changes.
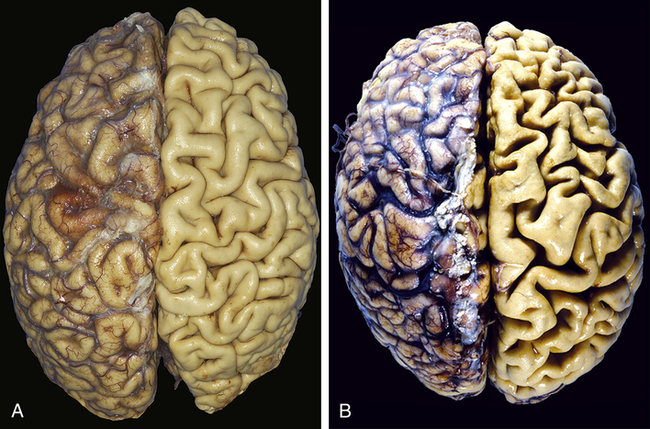
A, Normal brain of a young adult. B, Atrophy of the brain in an 82-year-old male with atherosclerotic disease. Atrophy of the brain is a result of aging and reduced blood supply. Note that loss of brain substance narrows the gyri and widens the sulci. The meninges have been stripped from the right half of each specimen to reveal the surface of the brain. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2007, Saunders.)
The atrophic muscle cell contains less endoplasmic reticulum and fewer mitochondria and myofilaments (part of the muscle fiber that controls contraction) than does the normal cell. In muscular atrophy caused by nerve loss, oxygen consumption and amino acid uptake are rapidly reduced. The mechanisms probably include decreased protein synthesis, increased protein catabolism, or both. Up-regulation of proteasome (protein degrading complex) activity is characteristic of atrophic muscle changes.1 The primary pathway of protein catabolism is the ubiquitin-proteasome pathway. Proteins degraded in this pathway are first conjugated to ubiquitin (another small protein) and then degraded by proteasomes.
Atrophy as a result of chronic malnutrition is often accompanied by a “self-eating” process called autophagy inducing autophagic vacuoles. These vacuoles are membrane-bound vesicles within the cell that contain cellular debris—small fragments of mitochondria and endoplasmic reticulum—and hydrolytic enzymes. Atrophic change causes a rapid increase in hydrolytic enzymes, which are isolated in autophagic vacuoles to prevent uncontrolled cellular destruction. Thus the vacuoles proliferate as needed to protect the uninjured organelles from the injured organelles and are eventually taken up and destroyed by lysosomes (see p. 7). Certain contents of the autophagic vacuole may resist destruction by lysosomal enzymes and persist in membrane-bound residual bodies. An example of this is granules that contain lipofuscin, the yellow-brown age pigment. Lipofuscin accumulates primarily in liver cells, myocardial cells, and atrophic cells.
Hypertrophy
Initial enlargement of the heart is caused by dilation of the cardiac chambers, is short-lived, and is followed by increased synthesis of cardiac muscle proteins, allowing muscle fibers to do more work. The nucleus also is hypertrophic and exhibits increased synthesis of deoxyribonucleic acid (DNA). The increase in cellular size is associated with an increased accumulation of protein in the cellular components (plasma membrane, endoplasmic reticulum, myofilaments, mitochondria) and not with an increase in the amount of cellular fluid. With time cardiac hypertrophy is characterized by extracellular matrix remodeling and increased growth of adult myocytes. The myocytes progressively increase in size and reach a limit beyond which no further hypertrophy can occur.2 Although hypertrophy can be classified as physiologic or pathologic, time may be the critical factor or determinant of the transition from physiologic to pathologic cardiac hypertrophy. Pathologic hypertrophy in the heart is secondary to hypertension or valvular dysfunction. Eventually, however, advanced hypertrophy can lead to myocardial failure (Figure 2-3), suggesting that restrictions in myocyte growth are critical determinants of ventricular dysfunction2 (see Chapter 32).
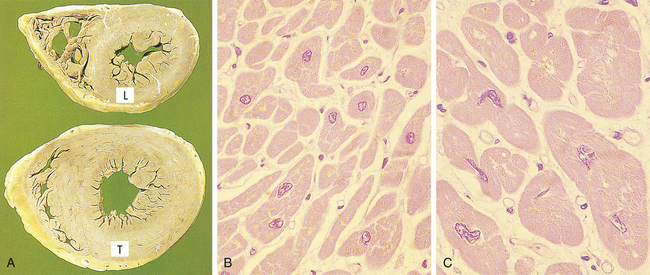
A, Transverse slices of a normal heart and a heart with hypertrophy of the left ventricle. (L, Normal thickness of left ventricular wall; T, thickened wall from heart in which severe narrowing of aortic valve caused resistance to systolic ventricular emptying.) B, Histology of cardiac muscle from a normal heart. C, Histology of cardiac muscle from a hypertrophied heart. (From Stevens A, Lowe JS, Scott I: Core pathology, ed 3, London, 2009, Mosby.)
Data for understanding myocyte hypertrophy have advanced; however, the reason the hypertrophied heart ultimately fails is not known. The molecular correlates that define decompensated (pathologic) hypertrophy remain obscure.2 The fetal genes present in embryonic development, α-skeletal actin and β-myosin heavy chains, have been viewed as the hallmark of pathologic hypertrophy.3
Historically it was thought that myocardial cells could not adapt to increased metabolic demands by mitotic division and production of new cells to share the work. This is now being challenged (see What’s New? Myocardial Hypertrophy, Stem Cells, Regeneration Controversy).
Hyperplasia
Loss of epithelial cells and cells of the liver and kidney triggers DNA synthesis and mitotic division. Increased cell growth is a multistep process involving the production of growth factors, which stimulate the remaining cells to synthesize new cell components and, ultimately, to divide. Hyperplasia and hypertrophy often occur together, although the specific mechanism is unknown. Both hyperplasia and hypertrophy take place if the cells are capable of synthesizing DNA.
Not all types of mature cells have the same capacity for compensatory hyperplastic growth. Nondividing tissues contain cells that can no longer (i.e., postnatally) go through the cell cycle and undergo mitotic division. These cells include neurons and skeletal and cardiac muscle cells. Destruction of neurons in the central nervous system is usually replaced by proliferation of the glial cells, the supportive structures. Recent data, however, have demonstrated neuron development from stem cells in adult brains.4 Mature skeletal muscle cells do not divide but have been shown to have regenerative capacity from differentiation of satellite cells that are part of the endomysial sheaths. Additionally, large injury to cardiac muscle cells is followed by scar formation. However, laboratory experiments show cardiac muscle cells may have limited regenerative capacity.
Significant compensatory hyperplasia occurs in epidermal and intestinal epithelia, hepatocytes, bone marrow cells, and fibroblasts, and some hyperplasia is noted in bone, cartilage, and smooth muscle cells. An example of compensatory hyperplasia is a callus, or thickening, of the skin as a result of hyperplasia of epidermal cells in response to a mechanical stimulus. Another example is the response to wound healing as part of the inflammation process (see Chapter 7).
Hormonal hyperplasia occurs chiefly in estrogen-dependent organs, such as the uterus and breast. After ovulation, for example, estrogen stimulates the endometrium to grow and thicken for reception of the fertilized ovum. If pregnancy occurs, hormonal hyperplasia, as well as hypertrophy, enables the uterus to enlarge. (Hormone function is described in Chapters 21 and 22.)
Pathologic hyperplasia is the abnormal proliferation of normal cells and can occur as a response to excessive hormonal stimulation or the effects of growth factors on target cells (Figure 2-4). Hyperplastic cells are identified by pronounced enlargement of the nucleus, clumping of chromatin, and the presence of one or more enlarged nucleoli. The most common example is pathologic hyperplasia of the endometrium, which is caused by an imbalance between estrogen and progesterone secretion with oversecretion of estrogen (see Chapter 24). Pathologic endometrial hyperplasia, which causes excessive menstrual bleeding, is under the influence of regular growth-inhibition controls. If these controls fail, hyperplastic endometrial cells can undergo malignant transformation. (Malignant cell transformation is discussed in Chapter 12.)
Dysplasia: Not a True Adaptive Change
Dysplasia is often classified as mild, moderate, or severe; however, this subjective scheme has prompted recommendations to use either “low grade” or “high grade,” for example, of the female reproductive tract (i.e., Papanicolaou [Pap] test [discussed in Chapter 24]) (Figure 2-5). Data indicate that atypical hyperplasia is a strong predictor of breast cancer development.5,6 If the inciting stimulus is removed, dysplastic changes often are reversible.
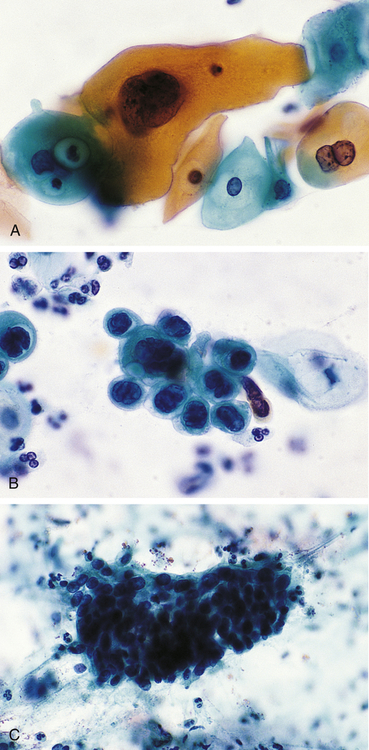
A, Mild dysplasia. B, Severe dysplasia. C, Carcinoma in situ (see Chapter 12). (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Metaplasia
Metaplasia is the reversible replacement of one mature cell by another, sometimes less differentiated, cell type. The best example of metaplasia is replacement of normal columnar ciliated epithelial cells of the bronchial (airway) lining by stratified squamous epithelial cells (Figure 2-6). The newly formed squamous epithelial cells do not secrete mucus or have cilia, causing loss of a vital protective mechanism.
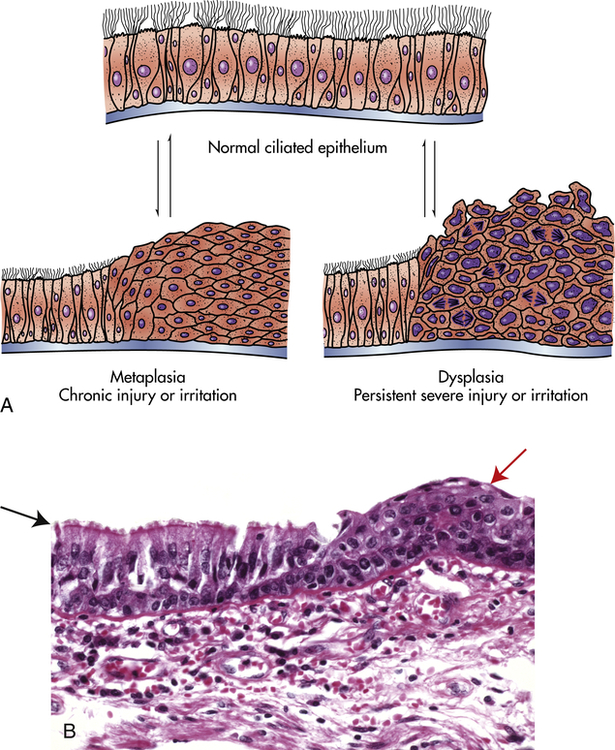
A, Normal ciliated epithelium, metaplasia, and dysplasia. B, Histologic slide with upper left (black arrow) normal columnar epithelium and basement membrane, and upper right (red arrow) squamous metaplasia. (B from Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2007, Saunders.)
Cellular Injury
Injury to cells and to extracellular matrix (ECM) leads to injury of tissues and organs ultimately determining the structural patterns of disease. Loss of function derives from cell and ECM injury and cell death. Cellular injury occurs if the cell is unable to maintain homeostasis—a normal or adaptive steady state—in the face of injurious stimuli or stress. Injured cells may recover (reversible injury) or die (irreversible injury). Injurious stimuli include chemical agents, lack of sufficient oxygen (hypoxia), free radicals, infectious agents, physical and mechanical factors, immunologic reactions, genetic factors, and nutritional imbalances. Types of cellular injury and their responses are summarized in Table 2-1 and Figure 2-7.
TABLE 2-1
PROGRESSIVE TYPES OF CELL INJURY AND RESPONSES
| TYPE | RESPONSES |
| Adaptation | Atrophy, hypertrophy, hyperplasia, metaplasia |
| Active cell injury | Immediate response of “entire” cell |
| Reversible | Loss of adenosine triphosphate (ATP), swelling of cell, detachment of ribosomes, autophagy of lysosomes |
| Irreversible | “Point of no return” structurally when severe vacuolization of mitochondria occurs and Ca++ moves into the cell, including mitochondrial membrane damage |
| Necrosis | Common type of cell death with severe cell swelling and breakdown of organelles |
| Apoptosis, a type of programmed cell death | Cellular self-destruction for elimination of unwanted cell populations |
| Chronic cell injury (subcellular alterations) | Persistent stimuli response may involve only specific organelles or cytoskeleton (e.g., phagocytosis of bacteria) |
| Accumulations or infiltrations | Water, pigments, lipids, glycogen, proteins |
| Pathologic calcification | Dystrophic and metastatic calcification |
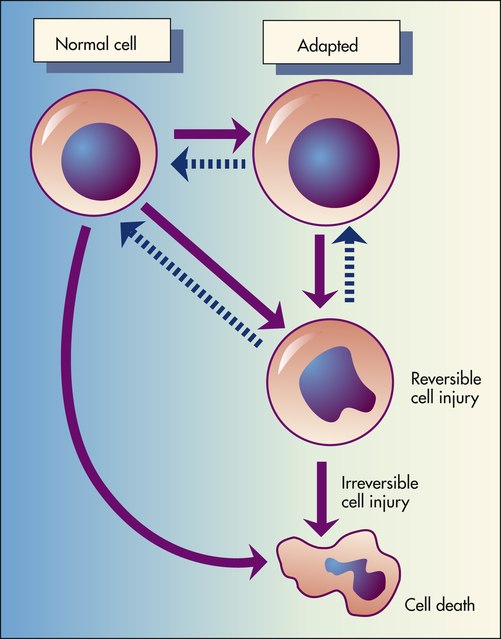
Depicted here is the relationship among normal, adapted (hypertrophy), and reversibly injured cells and cell death of myocardial cells.
Cell injury and cell death often result from exposure to toxic chemicals, infections, and hypoxia. (Infections are discussed in Chapter 10.) The mechanisms causing chemical and hypoxic injury are perhaps the best understood. Both of these mechanisms can lead to disruption of selective permeability (i.e., transport mechanisms) of the plasma membrane; reduction or cessation of cellular metabolism; lack of protein synthesis; damage to lysosomal membranes with leakage of destructive enzymes into the cytoplasm; enzymatic destruction of cellular organelles; cellular death (exhibited by nuclear changes); and phagocytosis of the dead cell by cellular components of the acute inflammatory response (see Chapter 7). The extent of cellular injury depends on the type, state (including level of cell differentiation and increased susceptibility to fully differentiated cells), and adaptive processes of the cell, as well as the type, severity, and duration of the injurious stimulus. Two individuals exposed to an identical stimulus may incur varying degrees of cellular injury. Modifying factors, such as nutritional status, can profoundly influence the extent of injury. The precise “point of no return” that leads to cellular death is a biochemical puzzle, and the exact mechanisms responsible for the transition from reversible to irreversible cellular damage are being debated.
General Mechanisms of Cell Injury
Cells are complex units, and therefore the mechanisms responsible for cell injury leading to necrotic cell death are numerous and interrelated and depend on a delicate balance between intracellular and extracellular events. There are, however, common biochemical themes important to cell injury and cell death regardless of the injuring agent (Table 2-2). Examples of cell injury are: (1) hypoxic injury, (2) reactive oxygen species and free radical–induced injury, and (3) chemical injury.
TABLE 2-2
COMMON THEMES IN CELL INJURY AND CELL DEATH
| THEME | COMMENTS |
| ATP depletion | Loss of mitochondrial ATP and decreased ATP synthesis; results include cellular swelling, decreased protein synthesis, decreased membrane transport, and lipogenesis, all changes that contribute to loss of integrity of plasma membrane (see text) |
| Oxygen and oxygen-derived free radicals | Lack of oxygen is key in progression of cell injury in ischemia (reduced blood supply); activated oxygen species (free radicals, H2O2, 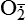 , NO) cause destruction of cell membranes and cell structure , NO) cause destruction of cell membranes and cell structure |
| Intracellular calcium and loss of calcium steady state | Normally intracellular cytosolic calcium concentrations are very low; ischemia and certain chemicals cause an increase in cytosolic Ca++ concentrations; sustained levels of Ca++ continue to increase with damage to plasma membrane; Ca++ causes intracellular damage by activating a number of enzymes (see text) |
| Defects in membrane permeability | Early loss of selective membrane permeability found in all forms of cell injury (see text) |

Hypoxic Injury
Hypoxia, or lack of sufficient oxygen, is the single most common cause of cellular injury (Figure 2-8). Hypoxia can result from a reduced amount of oxygen in the air, loss of hemoglobin or hemoglobin function, decreased production of red blood cells, consequences of respiratory and cardiovascular system diseases, and poisoning of the oxidative enzymes (cytochromes) within the cells. The most common cause of hypoxia is ischemia (reduced blood supply). Hypoxia can induce inflammation and inflamed lesions can become hypoxic (Figure 2-9).
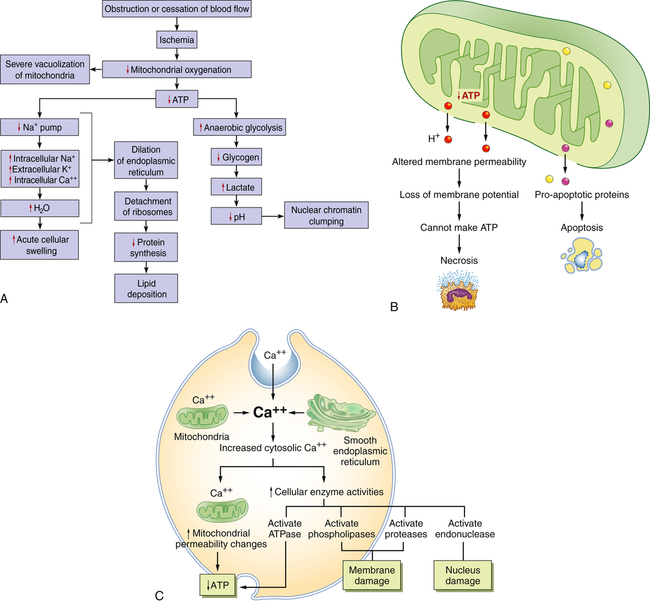
A, Consequences of decreased oxygen delivery or ischemia with decreased ATP. The structural and physiologic changes are reversible if oxygen is delivered quickly. Significant decreases in ATP concentration result in cell death, mostly by necrosis. B, Mitochondrial damage can result in changes in membrane permeability, loss of membrane potential, and decreased ATP. Between the outer and inner membranes of the mitochondria are proteins that can activate the cell’s suicide pathways, called apoptosis. C, Calcium ions are critical mediators of cell injury. Calcium ions are usually maintained at low concentrations in the cell’s cytoplasm; thus ischemia and certain toxins can initially cause an increase in the release of Ca++ from intracellular stores and later an increased movement (influx) across the plasma membrane.
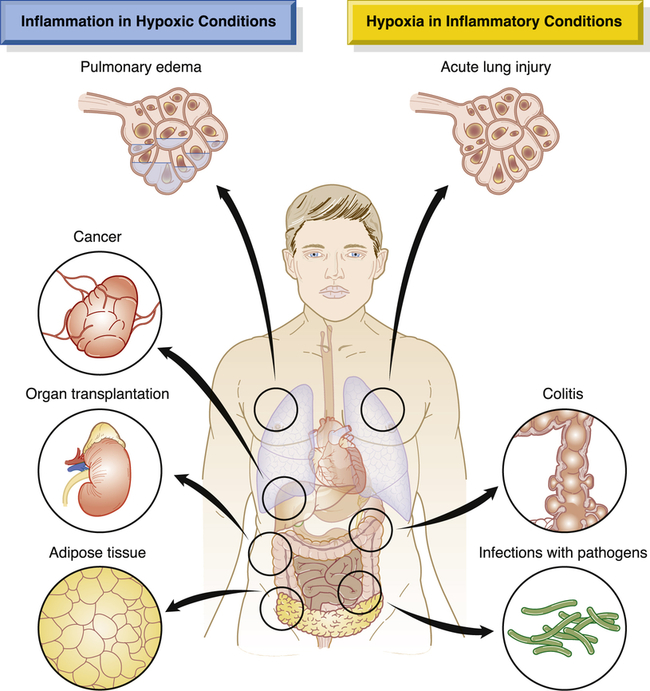
Shown is a simplified drawing of clinical conditions characterized by tissue hypoxia that causes inflammatory changes (left) and inflammatory diseases that ultimately lead to hypoxia (right). These diseases and conditions are discussed in more detail in their respective chapters. (Adapted from Eltzschig HK, Carmeliet P: N Engl J Med 364:656–665, 2011.)
Ischemic injury is often caused by gradual narrowing of arteries (arteriosclerosis) and complete blockage by blood clots (thrombosis). Progressive hypoxia caused by gradual arterial obstruction is better tolerated than the sudden acute anoxia (total lack of oxygen) caused by a sudden obstruction, such as can occur with an embolus (a blood clot or other plug in the circulation). An acute obstruction in a coronary artery can cause myocardial cell death (infarction) within minutes if the blood supply is not restored, whereas the gradual onset of ischemia usually results in myocardial adaptation. Myocardial infarction and stroke, which are common causes of death in the United States, generally result from atherosclerosis (a type of arteriosclerosis) and consequent ischemic injury. (Vascular obstruction is discussed in Chapter 32.)
A reduction in ATP levels causes the plasma membrane’s sodium-potassium (Na+-K+) pump and sodium-calcium exchange to fail, which leads to an intracellular accumulation of sodium and calcium, resulting in cellular swelling and diffusion of potassium out of the cell. (The Na+-K+ pump is discussed in Chapter 1.) Because all cells are bathed in a fluid rich in calcium ions, cell membrane damage allows rapid movement of calcium intracellularly. The movement of water and ions into the cell causes early dilation of the endoplasmic reticulum. Dilation causes the ribosomes to detach from the rough endoplasmic reticulum, resulting in reduced protein synthesis. With continued hypoxia, the entire cell becomes markedly swollen, with increased concentrations of sodium, water, and chloride and decreased concentrations of potassium. These disruptions are reversible if oxygen is restored. If oxygen is not restored, however, there is vacuolation (formation of vacuoles or cytoplasmic small cavities) within the cytoplasm, swelling of lysosomes, and marked swelling of the mitochondria resulting from mitochondrial membrane damage. Continued hypoxic injury with accumulation of calcium subsequently activates multiple enzyme systems, including proteases, nitric oxide synthase, phospholipases, and endonuclease, resulting in cytoskeleton disruption, membrane damage, activation of inflammation, DNA and chromatin degradation, ATP depletion, and eventual cell death (see Figures 2-8 and 2-23 [p. 86]). Structurally, with plasma membrane damage, extracellular calcium readily moves into the cell and intracellular calcium stores are released.
Intracellular calcium results in the activation of enzymes that can further damage membranes, proteins, ATP, and nucleic acids. The increased permeability of the membrane causes continued loss of proteins, essential coenzymes, and ribonucleic acids. In addition, the substrates necessary to reconstitute ATP are lost. Increased intracellular calcium levels activate cell enzymes (caspases) that promote cell death by apoptosis (see Figure 2-26 [p. 89]). Continued ischemia causes irreversible injury that is associated structurally with severe swelling of the mitochondria, severe damage to plasma membranes, and swelling of lysosomes.
Restoration of oxygen, however, can cause additional injury called reperfusion (reoxygenation) injury. Reperfusion injury results from the generation of highly reactive oxygen intermediates (oxidative stress), including hydroxyl radical (OH•), superoxide  , and hydrogen peroxide (H2O2) (see p. 59). These radicals can all cause further membrane damage and mitochondrial calcium overload. The white blood cells (neutrophils) are especially affected with reperfusion injury, including neutrophil adhesion to the endothelium.
, and hydrogen peroxide (H2O2) (see p. 59). These radicals can all cause further membrane damage and mitochondrial calcium overload. The white blood cells (neutrophils) are especially affected with reperfusion injury, including neutrophil adhesion to the endothelium.
Reperfusion is a serious complication and an important mechanism of injury in instances of tissue transplantation and in myocardial, hepatic, intestinal, cerebral, renal, and other ischemic syndromes, including stroke.7 Xanthine dehydrogenase, an enzyme that normally uses oxidized nicotinamide adenine dinucleotide (NAD+) as an electron acceptor, is converted during reperfusion with oxygen to xanthine oxidase. During the ischemic period, excessive ATP consumption leads to the accumulation of the purine catabolites hypoxanthine and
xanthine, which upon subsequent reperfusion and influx of oxygen are metabolized by xanthine oxidase to make massive amounts of superoxide and hydrogen peroxide. These radicals can all cause membrane damage and mitochondrial calcium overload.3 Cardiac ischemia and reperfusion injury cause excessive reactive oxygen species (ROS) and calcium overload of the mitochondria. These changes presumably lead to the opening of a large conductance pore on the mitochondrial membrane called the mitochondrial permeability transition pore (MPTP) with massive escape of ATP and solutes leading to cell death activation (apoptosis).8 Cardioprotection from ischemia/reperfusion injury is an important focus of much research (see What’s New? Cardioprotection for Ischemia-Reperfusion Injury). Other potential and current treatments include use of antioxidants, blockage of inflammatory mediators, and inhibition of apoptotic pathways.
Free Radicals and Reactive Oxygen Species—Oxidative Stress
An important mechanism of membrane damage is injury induced by free radicals, especially by excess ROS called oxidative stress (Figure 2-10). Oxidative stress occurs when excess ROS overwhelms endogenous antioxidant systems (Box 2-1). A free radical is an electrically uncharged atom or group of atoms having an unpaired electron. Having one unpaired electron makes the molecule unstable; thus to stabilize, it gives up an electron to another molecule or steals one. Therefore, it is capable of injurious chemical bond formation with proteins, lipids, and carbohydrates—key molecules in membranes and nucleic acids. Free radicals are difficult to control and initiate chain reactions. Emerging data indicate that ROS play major roles in the initiation and progression of cardiovascular alterations associated with hypertension, hyperlipidemia, diabetes mellitus, ischemic heart disease, chronic heart failure, and sleep apnea.9,10 ROS generation is thought to lead to vascular endothelial injury and consequently atherosclerosis. Up-regulation of adhesion molecule production in the endothelium can be accomplished by ROS, which diminishes nitric oxide (NO) synthase activity and promotes NO breakdown. This disturbance of the vascular environment presumably causes a reduction of endothelial-dependent vasodilation.11 Such reduction in endothelium-dependent vasodilation has been demonstrated through intra-arterial infusion of vasoactive agents. Specific mechanisms by which vascular endothelial dysfunction may lead to adverse cardiovascular events include vasoconstriction, vascular smooth muscle proliferation, hypercoagulability, and thrombosis.12
BOX 2-1 ROS AND PROLIFERATION, APOPTOSIS, AND NECROSIS
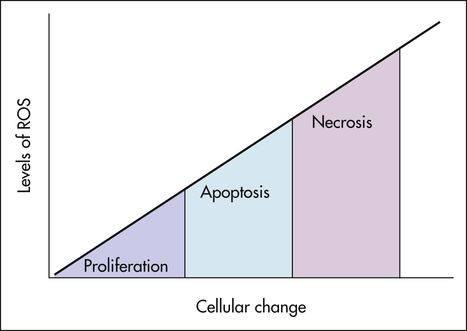
Data from Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004; Valko M et al: Int J Biochem Cell Biol 39(1):44–84, 2007.
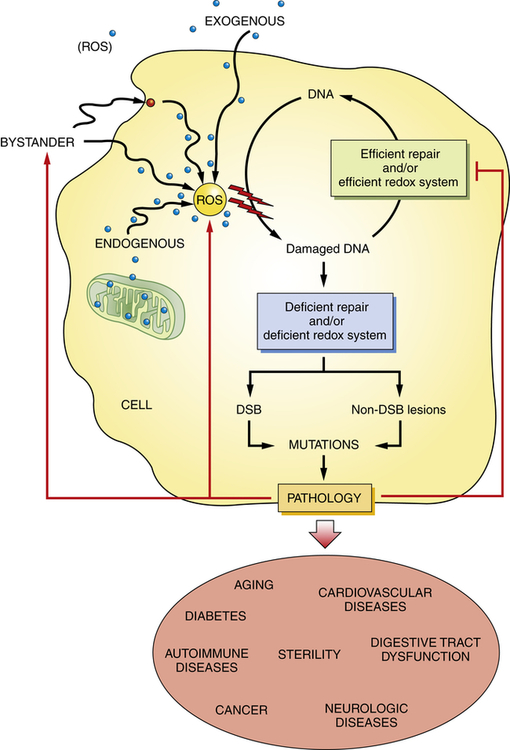
Reactive oxygen species (ROS) have a role in a wide variety of diseases and age-related diseases including cancer, neurologic disease, type 2 diabetes, autoimmune and cardiovascular diseases, infertility, and normal aging. Chronic exposure to ROS and decreased DNA repair can result in persistent DNA mutations. Accumulation of DNA lesions can lead to DNA double strand breaks (DSB) and disease onset/progression. Diseased cells can in turn develop ROS and decrease the efficiency of the DNA repair mechanism. (Adapted from Sedelnikova OA et al: Mutat Res 704:152–159, 2010.)
ROS produced by migrating inflammatory cells (e.g., neutrophils), as well as vascular cells (endothelial cells, vascular smooth muscle cells, and adventitial fibroblasts), have distinct effects on each cell type.13 These cell effects are shown in Figure 2-11. When inflammatory responses also are activated, there is consequent activation of endothelial cells, leukocytes, and platelets. These activated cells express adhesion molecules and proinflammatory cytokines that may further exacerbate inflammatory responses and cause endothelial cell injury and dysfunction, promoting the development of cardiovascular morbidities.14
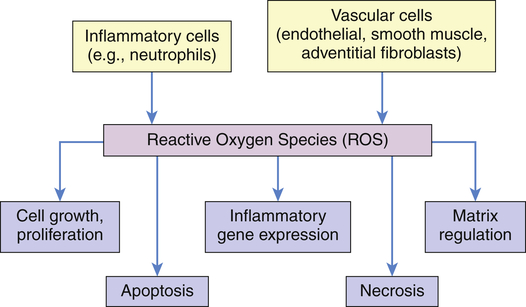
All cells are capable of making reactive oxygen species (ROS). Emphasis has been on inflammatory and vascular cells because of their widespread disease-causing effect. Some examples include angiotensin II, which can induce vascular smooth muscle cells (VSMCs) to hypertrophy; NAD(P)H oxidase–derived ROS has been implicated in the growth response; H2O2 has been shown to induce proliferation and migration of endothelial cells; ROS act as mediators of vascular endothelial growth factor, thus modulating angiogenesis; endothelial injury or exposure to
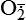 and H2O2 induces apoptosis of endothelial cells. Activity of the extracellular matrix by matrix metalloproteinases (MMPs) can be modulated by ROS. Cytokines play a significant role in the progression of vascular lesions. An important mechanism by which cytokine gene expression is increased is the activation of nuclear factor–κβ (NF-κβ). NF-κβ is a ROS-sensitive transcription factor and has a role in the expression of proinflammatory genes. (Data from Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004; Dröge W: Physiol Rev 82:47–95, 2002; Valko M et al: Int J Biochem Cell Biol 39[1]:44–84, 2007.) (Also see Box 2-1.)
and H2O2 induces apoptosis of endothelial cells. Activity of the extracellular matrix by matrix metalloproteinases (MMPs) can be modulated by ROS. Cytokines play a significant role in the progression of vascular lesions. An important mechanism by which cytokine gene expression is increased is the activation of nuclear factor–κβ (NF-κβ). NF-κβ is a ROS-sensitive transcription factor and has a role in the expression of proinflammatory genes. (Data from Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004; Dröge W: Physiol Rev 82:47–95, 2002; Valko M et al: Int J Biochem Cell Biol 39[1]:44–84, 2007.) (Also see Box 2-1.)Free radicals may be initiated within cells by (1) the absorption of extreme energy sources (e.g., ultraviolet light, x-rays); (2) the occurrence of endogenous reactions, such as redox reactions in which oxygen is reduced to water, as evident in systems involved in electron and oxygen transport (all biologic membranes contain redox systems important for cellular defense, for example, inflammation, iron uptake, growth and proliferation, and signal transduction) (Figure 2-12); or (3) the enzymatic metabolism of exogenous chemicals or drugs (e.g., chloromethyl [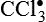 ], a product of carbon tetrachloride [CCl4]). Table 2-3 describes the most significant free radicals.
], a product of carbon tetrachloride [CCl4]). Table 2-3 describes the most significant free radicals.
TABLE 2-3
BIOLOGICALLY RELEVANT FREE RADICALS
| FREE RADICAL | COMMENTS |
| Reactive oxygen species (ROS) Superoxide 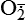 O2→ oxidase 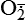 | Generated either (1) directly during autooxidation in mitochondria, or (2) enzymatically by enzymes in the cytoplasm, such as xanthine oxidase or cytochrome P-450; once produced, it can be inactivated spontaneously or more rapidly by the enzyme superoxide dismutase (SOD): 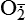 + + 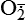 + 2H+ → SOD H2O2 + + 2H+ → SOD H2O2 + 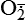 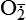 , a signaling molecule in growing or differentiating tissue, including hypertrophy, can alter cellular responses to growth factors and vasoconstrictor hormones; increasing levels of , a signaling molecule in growing or differentiating tissue, including hypertrophy, can alter cellular responses to growth factors and vasoconstrictor hormones; increasing levels of 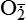 may lead to apoptosis (see Figure 2-11) may lead to apoptosis (see Figure 2-11) |
Hydrogen peroxide (H2O2)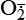 + + 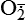 + 2H → SOD H2O2 + O2 + 2H → SOD H2O2 + O2or Oxidases present in peroxisomes O2 peroxisome 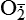 → SOD H2O2 → SOD H2O2 | Generated by SOD or directly by oxidases in intracellular peroxisomes; SOD is considered an antioxidant because it converts superoxide to 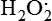 , catalase (another antioxidant) can then decompose H2O2 to O2 + H2O; H2O2 can serve as a cellular signaling molecule , catalase (another antioxidant) can then decompose H2O2 to O2 + H2O; H2O2 can serve as a cellular signaling molecule |
| Hydroxyl radicals (OH•) H2O → H• + OH• or Fe++ + H2O2 → Fe+++ + OH• + OH− or H2O2 + 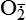 → OH• + OH− + O2 → OH• + OH− + O2 | Generated by the hydrolysis of water caused by ionizing radiation or by interaction with metals—especially iron (Fe) and copper (Cu); iron is important in toxic oxygen injury because it is required for maximal oxidative cell damage; OH• is highly reactive and can modify cellular macromolecules and cause toxicity |
| Nitric oxide (NO) NO• + 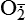 → ONOO− + H+ → ONOO− + H+↑ ↓ OH• + NO2 ONOOH → 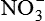 | NO by itself is an important mediator that can act as a free radical; it can be converted to another radical—peroxynitrite anion (ONOO−), as well as NO2• and 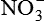 ; NO is formed in neuronal cells, where it modulates neurotransmission; in endothelial cells as a modulator of vessel relaxation; and in neutrophils and macrophages as a factor in vessel relaxation and inactivation of pathogens ; NO is formed in neuronal cells, where it modulates neurotransmission; in endothelial cells as a modulator of vessel relaxation; and in neutrophils and macrophages as a factor in vessel relaxation and inactivation of pathogens |

Data from Kumar V et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2010, Saunders; Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004.
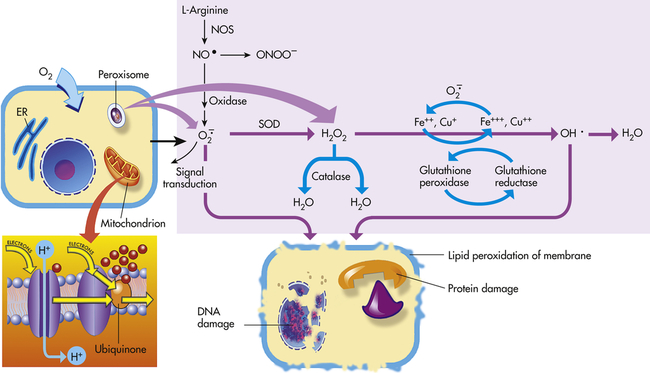
Mitochondria have four sites of entry for electrons coming into the electron-transport system: one for reduced nicotinamide adenine dinucleotide (NADH) and three for the reduced form of flavin adenine dinucleotide (FADH2). These pathways meet at the small, lipophilic molecule ubiquinone (coenzyme Q), at the beginning of the common electron-transport pathway. Ubiquinone transfers electrons in the inner membrane, ultimately enabling their interaction with O2 and H2 to yield H2O. In so doing, the transport allows free energy change and the synthesis of 1 mol of adenosine triphosphate (ATP). With the transport of electrons, free radicals are generated within the mitochondria. ROS (H2O2, OH•, and
 and nitric oxide [NO]) act as physiologic modulators of some mitochondrial functions but also may cause cell damage. O2 is converted to superoxide (
and nitric oxide [NO]) act as physiologic modulators of some mitochondrial functions but also may cause cell damage. O2 is converted to superoxide ( ) by oxidative enzymes in the mitochondria, endoplasmic reticulum (ER), plasma membrane, peroxisomes, and cytosol. O2 is converted to H2O2 by superoxide dismutase (SOD) and further to OH• by the Cu++/Fe++ Fenton reaction. Superoxide catalyzes the reduction of Fe++ to Fe+++, thus increasing OH• formation by the Fenton reaction. H2O2 is also derived from oxidases in peroxisomes. The NO• (radical) is produced by the oxidation of one of the terminal guanido-nitrogen atoms of l-arginine. Depending on the microenvironment, NO can be converted to other reactive nitrogen species including the highly reactive peroxynitrate (ONOO−). Both OH• and ONOO− are very reactive and can modify cellular macromolecules and cause toxicity. The less reactive molecules
) by oxidative enzymes in the mitochondria, endoplasmic reticulum (ER), plasma membrane, peroxisomes, and cytosol. O2 is converted to H2O2 by superoxide dismutase (SOD) and further to OH• by the Cu++/Fe++ Fenton reaction. Superoxide catalyzes the reduction of Fe++ to Fe+++, thus increasing OH• formation by the Fenton reaction. H2O2 is also derived from oxidases in peroxisomes. The NO• (radical) is produced by the oxidation of one of the terminal guanido-nitrogen atoms of l-arginine. Depending on the microenvironment, NO can be converted to other reactive nitrogen species including the highly reactive peroxynitrate (ONOO−). Both OH• and ONOO− are very reactive and can modify cellular macromolecules and cause toxicity. The less reactive molecules 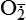 and H2O2 can serve as cellular signaling molecules. The major antioxidant enzymes include SOD, catalase, and glutathione peroxidase. (Data from Dröge W: Physiol Rev 82:47–95, 2002; Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004.)
and H2O2 can serve as cellular signaling molecules. The major antioxidant enzymes include SOD, catalase, and glutathione peroxidase. (Data from Dröge W: Physiol Rev 82:47–95, 2002; Buetler TM, Krauskopf A, Ruegg UT: News Physiol Sci 19:120–123, 2004.)Although wide-ranging effects can occur from these reactive species, three are particularly important in regard to cell injury: (1) peroxidation of lipids; (2) alterations of proteins causing fragmentation of polypeptide chains; and (3) alterations of DNA, including breakage of single strands. Lipid peroxidation is the destruction of unsaturated fatty acids. Fatty acids of lipids in membranes possess double bonds between some of the carbon atoms. Such bonds are vulnerable to attack by oxygen-derived free radicals, especially OH·. The lipid-radical interactions themselves yield peroxides. The peroxides instigate a chain reaction resulting in membrane, organelle, and cellular destruction. Because of the increased understanding of free radicals, a growing number of diseases and disorders have been linked either directly or indirectly to these reactive species (Box 2-2).



