Chapter 39
Alterations of Renal and Urinary Tract Function in Children

Structure and Function of the Urinary System in Children
Development of the Urinary System
The embryonic urinary system develops as three sets of sequentially replaced organs: the pronephros, mesonephros, and metanephros. The pronephros is a nonfunctional structure that arises at the level of the cervical and upper thoracic regions during the third fetal week and connects the primitive wolffian duct to the cloaca as the foundation for male sexual development (Figure 39-1). The development of the mesonephros and metanephros is described in Figure 39-1. The Wilms tumor 1 (WT1) gene plays an important role at all stages of kidney development and maintenance of kidney function. The wingless type signaling (WNT signaling) transduction pathway also is important for mesenchyme growth and differentiation.1,2
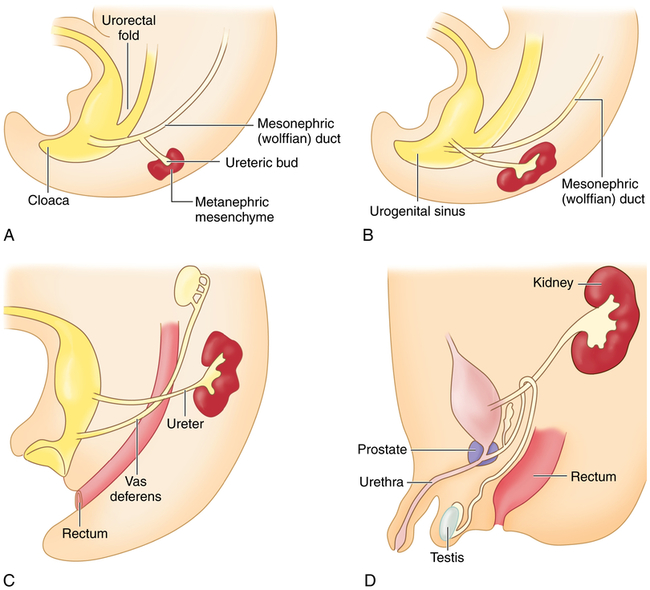
The mesonephros begins development more caudally about the fourth fetal week and begins excretory function in the sixth week. Most of the mesonephros degenerates and disappears by the end of the embryonic period. The metanephros, the permanent kidney, arises distal to the bifurcation of the aorta and develops from two different sources: (1) the ureteric bud (metanephric duct) forms as an outgrowth of the mesonephritic (wolffian) duct and grows dorsocranially and starts subdividing to become the collecting system for the kidneys by forming the ureter, renal pelvis, and calyces; by the fifth fetal month it will have progressively branched into the collecting ducts; and (2) the metanephrogenic mesenchyme sits atop the terminal branches of the collecting ducts and develops into primitive glomeruli and uriniferous tubules (A). Genetic information from the metanephrogenic mesenchyme guides the development of the ureteric bud. Establishing the connection between the uriniferous tubules and the collecting ducts is a vital part of kidney development; errors in this stage can result in polycystic kidneys. As the embryo grows, the definitive kidneys migrate from the caudal position to the lumbar region and the ureters connect with the bladder (B-D). In the 8-week male embryo, the wolffian duct begins to give rise to the epididymis, the seminal vesicles, and the caudal part of the vas deferens (C). The external genitalia develop between 8 and 16 weeks, and testicular descent begins in month 7 of gestation (D). (From Goldman L, Schafer AI: Goldman’s Cecil medicine, ed 24, Philadelphia, 2012, Saunders.)
Fluid and Electrolyte Balance in Children
Because the kidney develops from the center toward the periphery, renal distribution of blood flow during the newborn period is primarily to the renal medulla. The result is a preferential flow to the medullary nephrons, which have comparatively short loops at this stage of development. The combination of higher blood flow and shorter loops produces a more dilute urine-approximately 600 to 700 mOsm (compared with 800 to 1200 mOsm in adults) (see Chapter 3). The immature kidney is also less responsive to the actions of antidiuretic hormone (vasopressin) (see Figure 3-5).3 The dilute urine is accentuated by a low rate of urea excretion, which is necessary to establish the concentration gradient in the medulla. Urea excretion is low primarily because infants are in a high anabolic state and use their protein for growth.
Because of a high hydrogen ion concentration, limited ability to regulate the internal environment, and lowered osmotic pressure, the infant’s renal system has a narrow chemical safety margin. The immaturity and smaller surface area of the tubules also may diminish the water reabsorption response to antidiuretic hormone (ADH). An immature tubular transport capacity means that the ability to excrete a potassium load, reabsorb bicarbonate, or buffer hydrogen with ammonia does not become efficient until approximately 2 years of age. Consequently, any disturbance such as diarrhea, infection, fasting for diagnostic tests, or improper feeding can rapidly lead to severe acidosis and fluid imbalance because the infant can rapidly develop over- or underhydration, or edema (see Chapter 3).4
After birth the proportion of total body water to body weight does not change markedly. Considerable change occurs, however, in the location of that body water as the child matures (see Chapter 3). Compared to an adult, the percentage of extracellular fluid volume of the newborn infant is nearly double. Decrease in extracellular fluid volume occurs in two different periods of rapid growth-infancy and adolescence.
Alterations in Renal and Bladder Function in Children
Congenital Abnormalities
Congenital abnormalities of the kidney and urinary tract occur in about 1 out of 500 newborns.5 Structural abnormalities range from minor, nonpathologic, or easily correctable anomalies to those that are incompatible with life. For example, the kidneys may fail to ascend from the pelvis to the abdomen, causing ectopic kidneys-which usually function normally. The kidneys also may fuse in the midline as they ascend, causing a single U-shaped horseshoe kidney, with an incidence rate of 0.25 to 0.61 per 10,000 births.6 Approximately one third of individuals with horseshoe kidneys are asymptomatic, and the most common problems are hydronephrosis, infection, stone formation, and tumors.7 Collectively, structural anomalies of the renal system account for approximately 45% of cases of renal failure in children.
Some anomalies are obvious at birth, whereas others remain silent or become apparent in childhood. The following structural anomalies are commonly associated with urinary tract malformations8:
• Chromosomal disorders, especially trisomy 13 (Patau syndrome) and trisomy 18
• Absent abdominal muscles (prune-belly syndrome)
• Anomalies of the spinal cord and lower extremities
• Imperforate anus or genital deviation
• Positive family history of renal disease (e.g., hereditary nephritis or renal cystic disease)
Hypospadias
Hypospadias is a congenital condition in which the urethral meatus is located on the ventral side or undersurface of the penis. The meatus can be located anywhere on the glans, the penile shaft, the base of the penis, the penoscrotal junction, or the perineum (Figure 39-2). This is the most common anomaly of the penis and occurs in about 1 in 125 infant boys. The etiology is multifactorial and related to disruptions in male hormones, including testosterone biosynthesis defects, steroid 5α-reductase type 2 mutations, genes associated with penile and urethral development, hormones administered for in vitro fertilization, advanced maternal age, and other environmental factors.9 Chordee, or penile torsion, may accompany hypospadias. In chordee a shortage of skin on the ventral surface causes the penis to bend or to “bow” ventrally (Figure 39-3). Penile torsion is a counterclockwise twist of the penile shaft. Partial absence of the foreskin, inguinal hernia, and cryptorchidism (undescended testes; see Chapter 25) are associated with the anomaly.10
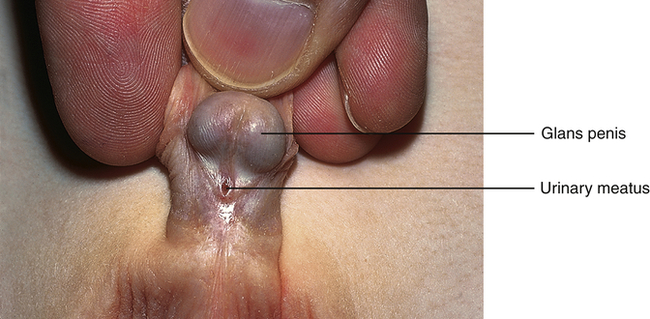
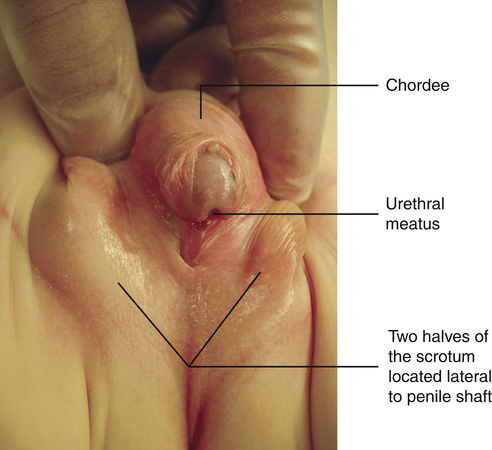
The goals for corrective surgery on the child with hypospadias are: (1) a straight penis when erect to facilitate sexual intercourse as an adult, (2) a uniform urethra of adequate caliber to prevent spraying during urinations, (3) a cosmetic appearance satisfactory to the individual, and (4) repair completed in as few procedures as possible. Formerly performed in two or more stages, hypospadias repairs are now done in one stage. Improvements in microsurgical techniques have enhanced outcomes and decreased complications and the need for follow-up surgery. Surgery is usually performed between 4 and 12 months of age.11
Epispadias
In boys the urethral opening may be small and situated behind the glans (anterior epispadias), or a fissure may extend the entire length of the penis and into the bladder neck (posterior epispadias). The majority of children with epispadias can achieve urinary continence, although surgical intervention may be necessary.12
Exstrophy of the Bladder
Exstrophy of the bladder is a rare extensive congenital anomaly of herniation of the bladder through the abdominal wall with failure of the abdominal muscles, pelvic ring, and pelvic floor musculature to fuse in the midline (Figure 39-4). The posterior portion of the bladder mucosa is exposed and appears bright red. The prevalence of exstrophy of the bladder is about 2.07 in 100,000 live births. Boys are predominant with a ratio of about 2:1 and this complication is more common in whites.13
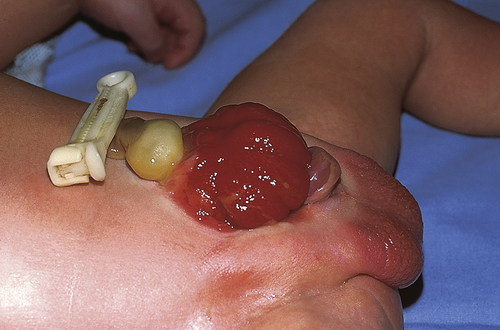
Exstrophy of the bladder is caused by intrauterine failure of the abdominal wall and the mesoderm of the anterior bladder to fuse. Urine seeps onto the abdominal wall from the ureters, causing a constant odor of urine and excoriation of the surrounding skin. The rectus muscles below the umbilicus are separated, and the pubic rami (bony projections of the pubic bone) are not joined. The clitoris in girls is divided into two halves with the urethra between them. The penis in boys is epispadic. In addition, the posterior aspect of the pelvis is externally rotated, which retroverts the acetabula and causes external rotation of the feet. This causes a waddling gait when the child first learns to walk, but most children quickly learn to compensate. Surgical intervention may be required.14 Because the exposed bladder mucosa becomes hyperemic and edematous, it bleeds easily and is painful. It should be covered with Silastic or a plastic dressing (e.g., kitchen plastic wrap) for protection from diaper irritation while permitting urine drainage. The unrepaired exstrophic bladder is cosmetically unacceptable and prone to cancerous changes as soon as 1 year after birth. Ideally the bladder and pubic defect should be closed before the infant is 72 hours old. Surgical reconstruction is usually performed within the first year either as a complete primary repair or as staged procedures. Staged procedures may include bladder augmentation and bladder neck and epispadias repair.15 Objectives of management include preservation of renal function, attainment of urinary control, prevention of infection, reconstructive repair of the defect, and improvement of sexual function and quality of life.
Ureteropelvic Junction Obstruction
Ureteropelvic junction (UPJ) obstruction is a blockage of the tapered point where the renal pelvis transitions into the ureter.16 An intrinsic malformation of smooth muscle hypertrophy and fibrosis produces obstruction in 90% of cases.17 It is the most common cause of hydronephrosis in neonates.18 Diagnosis is made by ultrasound. Open or endoscopic surgery to relieve the obstruction occurs if there is decline of renal drainage or function.19 During infancy or childhood, secondary ureteropelvic junction obstruction is caused by kinking or secondary scarring in the presence of high-grade vesicoureteral reflux. Children with UPJ obstruction have an increased risk of vesicoureteral reflux. Other defects are sometimes associated with ureteral duplication including complete ureteral duplication (abnormal growth of two ureters and ureteral orifices draining a single kidney), incomplete duplication (bifurcation of the ureter terminates into one ureteral orifice and serves a single kidney), and ureterocele (cystic dilation of the intravesical ureter). Obstruction of the distant ureter (ureterovesical junction obstruction) causes dilation of the entire ureter, renal pelvis, and calyceal system (megaureter). Megaureter can occur when a short acontractile segment of the ureter develops just above the ureterovesical junction or from ureteral reflux.20
Bladder Outlet Obstruction
Congenital causes of bladder outlet obstruction are rare and include urethral valves, urethral polyps, and urethral atresia. A urethral valve is a thin membrane that occludes the urethral lumen and obstructs urinary outflow in males. It is the most common cause of congenital lower urinary tract obstruction and renal failure. Most valves occur in the posterior urethra, although a few arise from the embryologically distinct anterior urethra.21 Urethral polyps arising from the prostatic urethra are rare. Symptoms of polyps may include hematuria; voiding issues, such as urinary retention or straining to void; and dilation of the upper tracts.22 Urethral atresia is absence of the urethra and is rare.
Congenital urethral valves or polyps can be diagnosed with prenatal ultrasound and treated with prenatal bladder shunting or with resection during the first days of life.23 Infants with significant renal (and pulmonary) hypoplasia who are unable to undergo primary resection may be managed with a vesicostomy, a small opening created between the bladder wall and the abdomen.24
Hypoplastic or Dysplastic Kidneys
During embryologic development the ureteric duct grows into the metanephric tissue, triggering the formation of the kidneys. If this growth does not occur, the kidney is absent-a condition called renal aplasia. A hypoplastic kidney is small with a decreased number of nephrons. These conditions may be unilateral or bilateral; the occurrence may be incidental or familial.24 Bilateral hypoplastic kidneys are a common cause of chronic renal failure in children. Segmental hypoplasia (the Ask-Upmark kidney) may be congenital or secondary to vesicoureteral reflux.25
Renal Agenesis
Renal agenesis (the absence of one or both kidneys) may be unilateral or bilateral, and may occur randomly or be hereditary. It may be an isolated entity or associated with anomalies in other organs.26
Unilateral renal agenesis occurs in approximately 1 of 1000 live births in the United States. Males are more often affected, and it is usually the left kidney that is absent. The single kidney is often completely normal so that the child can expect a normal, healthy life. The normal solitary kidney grows because of compensatory hypertrophy before27 and after birth. By the time the child is several years older, the volume of this kidney may approach twice the normal size. In some instances the single kidney is abnormally formed and associated with abnormalities of its collecting system.28 Extrarenal congenital abnormalities are relatively more common with unilateral renal agenesis.
Bilateral renal agenesis (also called Potter syndrome) is a rare disorder incompatible with extrauterine life.29 Approximately 75% of affected infants are male. Bilateral renal agenesis results from either an abnormal development of the normal progression from pronephros to mesonephros to metanephros or an isolated bilateral failure of development of the ureteral buds. The term Potter syndrome refers to the association with a specific group of facial anomalies (wide-set eyes, parrot-beak nose, low-set ears, and receding chin). Oligohydramnios (low amount of amniotic fluid) leads to underdeveloped lungs. Affected infants rarely live more than a few hours because of pulmonary insufficiency. Approximately 40% of affected infants are stillborn. Renal agenesis can be detected prenatally by ultrasound.
Polycystic Kidneys
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder that occurs in about 1 in 1000 live births in the United States. Mutations of two genes, PKD-1 (chromosome 16) and PKD-2 (chromosome 4), account for the disease most often presenting in late childhood or adulthood. The gene products (polycystins) regulate growth and differentiation of the tubular epithelium.30 Defects in the formation of epithelial cells and their cilium result in cyst formation and obstruction accompanied by destruction of renal parenchyma, interstitial fibrosis, and loss of functional nephrons. Cysts may occur in other organs including the liver, ovaries, and pancreas. Hypertension, heart valve defects, and cerebral and aortic aneurysms may develop. Urinary tract infection, hematuria, and flank pain may occur. Diagnosis is usually confirmed by ultrasound. Individuals may live for decades before developing symptoms of renal disease and not all progress to end-stage disease.31 Autosomal recessive PKD (ARPKD) with cystic changes in the kidney and liver is often first suspected on a prenatal ultrasound. The gene mutation for ARPKD (PKHD1) encodes a protein important to maintaining structural integrity and cellular function of the kidneys and liver. Renal replacement therapy is usually required during childhood or adolescence. Biomarkers and therapies to slow the progression of cyst development and renal failure are being studied.32
Glomerular Disorders
Glomerulonephritis
Acute glomerulonephritis includes a number of renal disorders in which proliferation and inflammation of the glomeruli are secondary to an immune mechanism (Table 39-1). (The major glomerulopathies and their histologic characteristics can be reviewed in Chapter 38 and Table 38-7.) The symptoms usually include the sudden onset of hematuria with red blood cell casts and proteinuria, and can be accompanied by renal salt and water retention, edema, hypertension, and in severe cases azotemia (i.e., decreased glomerular filtration rate). Chronic glomerulonephritis is the causative factor for 30% to 50% of renal failure in children and is the condition responsible for most school-age and teenage children requiring dialysis and kidney transplantation.
TABLE 39-1
PRIMARY GLOMERULONEPHRITIS IN CHILDREN
| CLASSIFICATION | FINDINGS |
| Cause | Poststreptococcal infection Related to other bacterial or viral infection Unknown |
| Immunologic mechanism | Antigen-antibody complex deposition Planted antigens with immune complex formed in situ Formation of antiglomerular basement membrane antibodies (rare) No immunologic cause established |
| Histopathology | No lesion Diffuse, focal, or segmented Membranous, proliferative, or combination of types Lobular, exudative, necrotizing, and other types Chronic with glomerular proliferation |
| Clinical manifestations of disease | Acute glomerulonephritis Persistent (chronic) glomerulonephritis Idiopathic nephrotic syndrome |
Acute Poststreptococcal Glomerulonephritis
Acute poststreptococcal glomerulonephritis (PSGN) is one of the most common immune complex-mediated renal diseases in children ages 5 to 15 years and it is representative of acute glomerulonephritis. It most commonly occurs after a throat (pharyngitis) or skin (impetigo) infection with nephritogenic strains of group A beta-hemolytic streptococci, although other bacteria (e.g., Staphylococcus) and viruses also may be responsible. Sporadic occurrences of infectious glomerulonephritis have been observed after bacterial endocarditis, which may be associated with streptococcal or staphylococcal microorganisms, or after viral diseases, such as varicella and hepatitides B and C. Glomerulonephritis develops with the deposition of antigen-antibody complexes (immunoglobulin G [IgG], IgA, and C3 complement) (see Chapter 8) in the glomerulus or the antigen may be trapped within the glomerulus and immune complexes formed in situ.33 The exact mechanism of immune complex formation is unknown. The immune complexes initiate inflammation and glomerular injury. Immunofluorescence microscopy shows lumpy deposits of immunoglobulin (IgG) and complement (C3) on the glomerular basement membrane (Figure 39-5). The thickened glomerular membrane contributes to decreased GFR. Activated complement, inflammatory cytokines, oxidants, proteases, and growth factors attack epithelial cells, alter membrane permeability, and cause hematuria and proteinuria. More severe renal disease is observed after a prolonged infection and before antibiotic therapy. Hypertension occurs primarily because of fluid retention.34
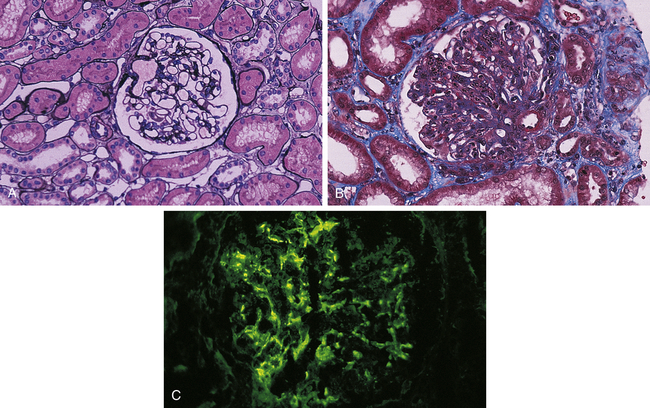
A, Normal glomerulus; note single-contoured walls, patent capillaries, inconspicuous mesangium, and degree of cellularity. (Periodic acid-methenamine silver stain.) B, Acute postinfectious glomerulonephritis. There is considerable increase in cellularity, mainly because of accumulation of numerous polymorphonuclear leukocytes in capillary lumina. Note numerous subepithelial hump-shaped fuchsinophilic deposits in many capillary walls. Protein precipitates (hyalinization) are in the arteriole. (Masson trichrome stain.) C, Postinfectious glomerulonephritis. Irregular mesangial and capillary wall immunostaining for C3. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Henoch-Schönlein Purpura Nephritis
Henoch-Schönlein purpura (HSP) nephritis (anaphylactoid purpura) is an immune-mediated IgA vasculitis that affects glomerular blood vessels, causing inflammation and damage to the vessel wall (see Chapter 38 for the pathophysiology of IgA nephropathy). The disease also involves small vessels in the skin and gut. Classic symptoms of HSP include palpable purpura, arthritis, abdominal pain, and renal disease characterized by gross or microscopic hematuria with mild or no proteinuria. Kidney biopsy demonstrates IgA deposition in the mesangium35 (see Table 38-7 in Chapter 38). The development of interstitial fibrosis and crescent formation from subepithelial immune deposits along the glomeruli increases the risk of chronic renal failure.36 Most children recover with supportive care, although some progress to kidney failure. Severe symptoms require corticosteroids and other immunosuppressant drugs.37
Hemolytic Uremic Syndrome
Pathophysiology
HUS has been associated with both bacterial and viral agents, as well as endotoxins, especially those from Escherichia coli 0157:H7 and recently Escherichia coli 0104:H4 (Shiga toxins).38 Potential sources of exposure include animals, unpasteurized beverages, and contaminated meat and vegetables. The disease also occurs with cancer and use of chemotherapeutic agents.39 In HUS, verotoxin (Shiga toxin) from E. coli is absorbed from the intestines into the blood, binds to polymorphonuclear leukocytes, and is transported to the kidney, causing a cascade of effects, including lysis of glomerular capillary endothelial cells, separation of endothelial cells from the basement membrane, activation and aggregation of platelets, and activation of the coagulation cascade. The glomerular arterioles become swollen and occluded with platelets and fibrin clots. There is decreased glomerular filtration, and the damaged glomerular membrane results in hematuria and proteinuria. Oliguria with renal failure occurs in up to 50% of children. Narrowed vessels damage passing erythrocytes. These damaged red blood cells, identified as burr cells, helmet cells, and fragmented red blood cells, are removed by the spleen, causing acute hemolytic anemia. Fibrinolysis, the process of dissolution of a clot, acts on precipitated fibrin, causing the fibrin split products to appear in serum and urine. The platelet clustering within damaged vessels, combined with the damage and removal of platelets, produces thrombocytopenia. Fibrin-rich thrombi can be found throughout the microcirculation.40 Other tissues, including the brain, liver, heart, and intestines, are often involved, which portends a poorer prognosis.
Evaluation and Treatment
Clinical evaluation includes history of preexisting illness, presenting symptoms, and urine and blood analysis. Antibiotics are not used in the initial treatment because they increase Shiga toxin release and increase the risk of HUS. Management consists of maintaining nutrition and hydration (to dilute toxins) and controlling hypertension, hyperkalemia, and seizures.41 When renal failure occurs, dialysis is indicated. Blood transfusions with packed red blood cells are needed to maintain reasonable hemoglobin levels. Most children recover; however, some will develop hypertension, proteinuria, or renal insufficiency or failure. Death usually occurs from complications related to CNS or myocardial involvement.42 Preventing Shiga toxin-producing bacterial infection (i.e., E. coli) prevents HUS.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


