Alterations in Immune Function
Faith Young Peterson
Key Questions
• How are hypersensitivity disorders detected, prevented, and treated?
• How do the etiologic processes of primary and secondary immune deficiency disorders differ?
• What are the clinical features of the common immunodeficiency disorders?

http://evolve.elsevier.com/Copstead/
The purposes of the immune system are to defend the body against invasion or infection by foreign substances called antigens, and to patrol for and destroy cells that are abnormal or damaged. Normally, the immune system works efficiently to accomplish these purposes, but in some situations inappropriate immune responses lead to disease.
These disorders can be divided into two general categories: (1) excessive immune responses and (2) deficient immune responses. The category of excessive immune responses includes disorders in which the immune system is overfunctioning or hyperfunctioning. Examples are autoimmunity and hypersensitivity disorders.
The category of deficient immune responses includes disorders in which the immune response is ineffective because of disease-causing genotypes or secondary/acquired dysfunction. Examples of deficient immune responses are severe combined immunodeficiency (SCID) syndrome, DiGeorge syndrome, and selective immunoglobulin A (IgA) deficiency. Human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) is a primary acquired immunodeficiency disorder that is discussed in Chapter 12. The secondary immunodeficiencies associated with white blood cell malignancies are included in Chapter 11.
Excessive Immune Responses
Excessive immune response disorders result from a functional increase in the activity of the immune system involving multiple, interacting immune cells. Autoimmunity and hypersensitivity are types of excessive immune response disorders. They are often related, and both may be present in patients with excessive immune responses. It may be helpful to think of autoimmunity as a way of describing the etiologic process, or cause, of abnormal excessive immune responses toward self tissues. Hypersensitivity disorders describe mechanisms of injury, or how the injury occurs, which may or may not involve autoimmunity. Autoimmunity is a general term that is used when the immune system attacks its own tissues. Most autoimmune reactions toward self tissues are mediated through type II (cytotoxic) and III (immune-complex) hypersensitivity mechanisms. For this reason, many autoimmune diseases also are considered hypersensitivity reactions. For example, myasthenia gravis is both an autoimmune disease and a type II hypersensitivity reaction. Immune complex glomerulonephritis is both an autoimmune disease and a type III hypersensitivity reaction. When hypersensitivity reactions occur in response to foreign antigens, such as bee venom, they are not autoimmune.
The causes of immune system overreactions are poorly understood. Interplay between genetic factors, including major histocompatibility complex (MHC) genes, and environmental factors is thought to be important in the development of autoimmune disorders. Most hypersensitivity disorders have familial tendencies also, but the specific genes and environmental agents remain to be discovered. Some evidence suggests that excessive immune responses may be the result of glucocorticoid resistance in target tissues. For example, the number of glucocorticoid receptors in circulating leukocytes is decreased 50% in patients with rheumatoid arthritis. This decrease in receptors would prevent adequate suppression of leukocyte activity and could further enhance inflammation.
Autoimmunity
Autoimmunity occurs when the immune system recognizes a person’s own cells (“self”) as foreign and mounts an immune response that injures self tissues. It is a failure of self tolerance. Identification and tolerance of self antigens occur during embryonic development. The adaptive immune response is the responsibility of antigen-specific T cells and B cells that learn to identify “self” in the thymus and bone marrow.1 The thymus gland is principally responsible for eliminating self-reactive cells.1 During this time, aggressive or intolerant self-reactive (autoreactive) lymphocytes are eliminated or suppressed (see Chapter 9). However, a small number of T cells escape thymic control, and move into the peripheral circulation. The immune system has other peripheral “checkpoints” to detect, limit, and control these “self” or “auto” reactive T cells.1 Autoimmune diseases result when self tolerance is lost and reactions between self antigens and the immune system occur causing dysregulation of proinflammatory and antiinflammatory mediators, cytokines, CD4+ and CD8+ T cells, B cells, and ubiquitin-editing enzyme A20 in dendritic cells.
Several theories have been proposed to explain how various immune system components and environmental triggers might interact to produce autoimmunity. However, no single theory can fully explain the loss of self tolerance that occurs in autoimmune diseases. A number of genetic and environmental factors interacting together contribute to the development of autoimmunity. The triggers for autoimmunity are not known exactly because autoimmune diseases are mediated by a variety of mechanisms, biochemical and cellular events, and responses to those events.
The theory of antigenic mimicry emphasizes the similarities between certain molecular segments of foreign antigens called epitopes and the person’s own cells. For example, all cells, whether self or foreign, are composed of proteins, carbohydrates, nucleic acids, and lipids. Certain viruses and bacteria evolve to look like “self” and use “molecular mimicry” to slip past the immune system defenses. Self cells with the same or similar molecular segments as these foreign epitopes can “fit” lymphocyte receptors. Therefore, these self antigens or autoantigens can be attacked as foreign under certain circumstances when the normal cell has been altered, such as by a viral infection that stimulates the immune response.2 The persistent presence of these autoantigens then acts as a constant source of stimulation to the immune system.2 A recent study of the molecular mimicry theory established an epidemiologic association between Campylobacter jejuni enteritis and the subsequent development of Guillain-Barré syndrome.2 This theory is also suggested as a cause of rheumatic heart disease attributable to cross-reactions between streptococcal antigens and human proteins.3 In both cases, autoantigens have been identified following exposure to bacterial or viral infection.
Another theory proposes that release of sequestered antigens triggers the autoimmune response. This theory suggests that certain self antigens are isolated from the immune system within an organ during the neonatal period. They are not in contact with antigen-processing cells during the embryonic period when self tolerance usually occurs. These hidden self antigens or sequestered proteins that are normally sheltered from immune recognition occur in sites such as the cornea of the eye, the testicles, or other areas not drained by lymphatics. If and when these sites are damaged later in life, the hidden or sequestered proteins are exposed to the immune system, which does not recognize them as self. Therefore, the damaged cells are attacked.
A number of T cell theories of autoimmunity have been proposed, including thymus gland defects, decreased suppressor T cell function, and altered T helper cells. The theories attributing autoimmunity to thymus gland defects state that maturation and differentiation of T cells are affected either by decreased hormone secretion or by failure of the thymus to expose T cells to all self products. The thymus gland is responsible for exposing developing T cells to self products produced in the thymus or carried to the thymus gland. If some self products are not exposed to the developing T cells, the product will not be recognized as self and will subsequently be attacked. However, recent evidence demonstrates that not all T cells maturate in the thymus. Thus not all T cells may experience or “learn” to recognize “self.”4 This lack of exposure to self products is thought to be a major factor in the development of generalized autoimmune diseases such as systemic lupus erythematosus (SLE). Thymus dysfunction in “programming” self-tolerance to pancreatic insulin-secreting islet β cells along with increased effector T cells and decreased regulatory T cells is thought to be the cause of autoimmune type 1 diabetes mellitus.5
The theory attributing autoimmunity to decreased or lost regulatory/suppressor T cell (Treg) activity states that decreased numbers of Treg cells fail to repress immunoglobulin activity. It is unclear in this theory if Treg cells are lost or if they are “reprogrammed” when exposed to inflammation.6 Some data suggest that Treg cells lose their forkhead box p3(FOXP3) protein and are transformed into effector T cells.6 It is also unclear if this disruption in the number of, or activity of, T cells may also be the result of genetic mutations. For example, a mutation of the transcription factor (FOXP3) causes impaired development and function of CD4+ T cells, which can lead to autoimmune inflammation. T helper cells 22 (Th22 cells) are a newly identified subset of T cells that are important mediators of chronic inflammation and autoimmunity by inducing the production of inflammatory cytokines (ll17a, ll17f, ll22, ll26) by CCR6+ cells.7
A number of B cell theories of autoimmunity also have been proposed. The theory attributing autoimmunity to escape of B cell tolerance proposes that certain B cells lose their responsiveness to suppressor T-cell messages. The B cell activation theories, which are well supported clinically and experimentally, suggest that extrinsic factors or intrinsic, genetic B cell defects cause autoantibody production and an increase in the number and activity of B cells. A number of extrinsic factors, including viruses, bacteria, antibiotics, proteolytic enzymes, and lipopolysaccharides, have been found to be B-cell activating factors that could trigger autoantibody production.
Research has linked mast cells to autoimmunity as well as hypersensitivity. Mast cells reside in most mucous membranes waiting for foreign proteins or bacteria to invade. Their cell membranes are studded with bacteria-sensing proteins, called Toll-like receptors, which cause release of up to 10,000 different chemicals in response to activation. Mast cells release the cytokine interleukin-1 (IL-1), attracting and enlisting inflammation-inducing cells to joints and leaking fluid into joints in the autoimmune disease of rheumatoid arthritis. They are also thought to be involved in development of irritable bowel syndrome and other functional gastrointestinal disorders.
Genetic Factors
Genetic predisposition seems to be an important factor in the development of autoimmune disorders. Gender, which is genetically determined, also influences the expression of autoimmune disorders. The exact mechanisms of gender and genetic influence on autoimmune expression have not been established, but the relationship is significant. Females are at significantly higher risk for developing autoimmunity compared to males. Different cytokine profiles can be associated with autoimmunity. Those with genetically low levels of tumor necrosis factor-α (TNF-α) and high levels of IL-10 may be more tolerant than those with normal levels.
The role of genetics is also supported by the observation that certain human major histocompatibility complex (MHC) genes located on chromosome 6p21 (also called human leukocyte antigen [HLA] genes) are frequently associated with certain autoimmune disorders (Table 10-1). The MHC gene region demonstrates a high level of polymorphism. One of the strongest correlations of MHC molecules with autoimmune disease is the linkage between the HLA-B27 phenotype and ankylosing spondylitis. In this case, 95% of all people with ankylosing spondylitis have a positive B27 phenotype. However, not everyone with a positive B27 phenotype develops ankylosing spondylitis, both because of differences in the way antigen is presented to the immune system and because of environmental factors. Other diseases are associated with different MHC phenotypes, but the correlation between risk for disease and presence of the disease marker is much lower. For example, Addison disease is associated with the HLA-DR3 phenotype, but it has only a 6% risk correlation. Juvenile rheumatoid arthritis is strongly associated with HLA-DR5.
TABLE 10-1
MAJOR HISTOCOMPATIBILITY GENES AND AUTOIMMUNE DISEASE
| DISEASE | HLA (MHC) ANTIGEN | FREQUENCY IN PATIENTS (%) | FREQUENCY IN CONTROLS (%) | RELATIVE RISK |
| Ankylosing spondylitis Caucasians Japanese | B27 B27 | 89 85 | 43 <1 | 69 207 |
| Rheumatoid arthritis | DR4 | 68 | 25 | 3.8 |
| Graves disease | Dw3 | 56 | 25 | 3.7 |
| Type 1 diabetes mellitus | DR3/DR4 heterozygous | 33 | ||
| Systemic lupus erythematosus | DR4 | 73 | 33 | 5 |
| Narcolepsy | DR2 | 100 | 34 | 358 |
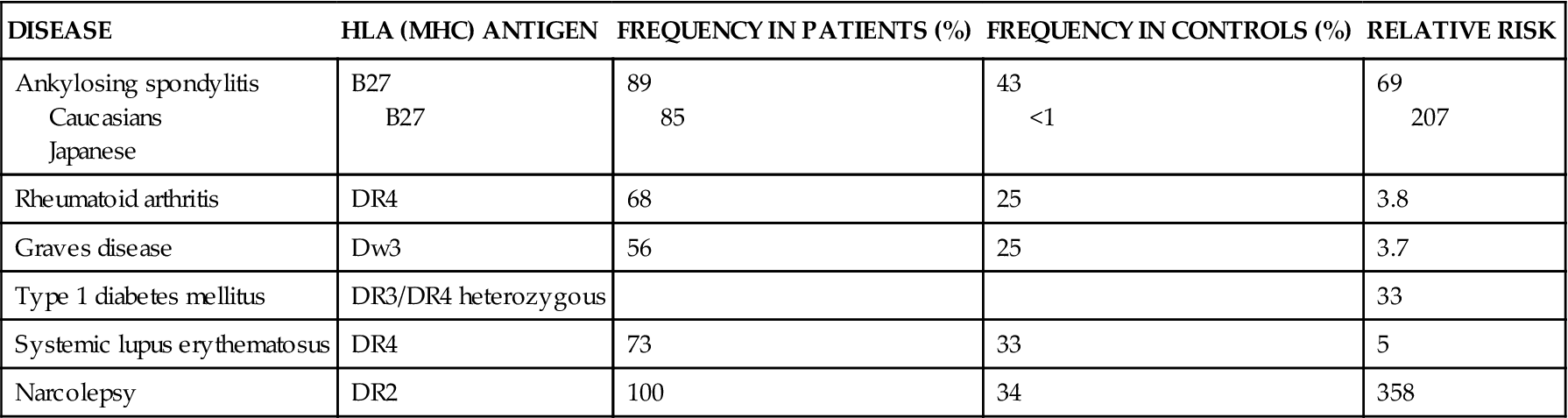
Data from Tierney LM, McPhee SJ, Papadakis MA, editors: Current medical diagnosis and treatment, ed 46, New York, 2007, Lange/McGraw-Hill.
There may also be disease-causing genotypes or genetic factors affecting expression of immune factors. For example, the lymphoid protein tyrosine phosphatase nonreceptor type 22 gene is associated with type 1 diabetes and other autoimmune disease. Tumor necrosis factor-α (TNF-α) is involved in acute and chronic inflammation, autoimmunity, and malignancies. Of special interest is the FOXP3 gene, which is expressed by CD4+ regulatory T cells. FOXP3 deficiency is associated with both primary immune deficiency disorders and autoimmune disorders owing to its effect on regulatory T-cell immune function.
Environmental Triggers
Chronic or multiple viral or bacterial infections may trigger the development of autoimmune disease in susceptible persons. Viruses can activate B cells, decrease the function of T cells, contribute to the development of antigenic mimicry, or insert viral components on cell surfaces and trigger immune reactions. For example, Epstein-Barr virus, cytomegalovirus, and bacteria such as Campylobacter jejuni and Helicobacter pylori have been frequently cited as potential triggers of autoimmune disease.
Environmental stress and occupational stress can affect the immune system because of their relationship to the neuroendocrine system, leading to inflammation or lymphokine release that activates T cells. Neuroendocrine and immune system interaction during life stress, such as shift work or workplace stress, promotes the synthesis and overproduction of proinflammatory cytokines. In genetically susceptible persons, this increase in systemic and local proinflammatory cytokines may affect the system’s balance enough to trigger autoimmune disease. There are also other linkages between the neuroendocrine and immune systems.
Although the etiology of autoimmunity continues to be investigated, the mechanisms whereby autoantibodies injure tissues are better understood. The autoantibodies produced by autoimmune disorders affect tissue by the mechanisms described for type II and type III hypersensitivity reactions found later in this chapter.
Pharmacotherapies
Immunosuppressive therapy is a common treatment for autoimmune disease. Because autoimmunity is expressed in different ways, the immunosuppressive treatment for each type of autoimmune disease is individualized, depending on disease expression. Immunosuppressive therapy, including corticosteroids and certain cytotoxic chemotherapeutic agents, has become an increasingly important treatment choice. These drugs are essential for inhibiting excessive or aberrant immune responses. The ideal immunosuppressive medication would be an agent that inhibits only the abnormal immune response without limiting the positive and protective functions of the immune system or causing any organ toxicity. Unfortunately, no immunosuppressive medication with these specific properties yet exists.
Immunosuppressive agents include corticosteroids, tumor necrosis factor inhibitors, immunomodulators, and cytotoxins. Corticosteroids decrease the number of lymphocytes and decrease antibody formation, as well as alter the functional activities of lymphocytes. They also have many other activities as a result of their glucocorticoid function. Corticosteroids tend to be used in the treatment of many autoimmune diseases and are the oldest of the immunosuppressive drugs. The adverse effects that occur during corticosteroid use often limit their extended use over time. The common side effects include hypercorticism with changes in fat distribution and buffalo hump formation, suppression of the HPA axis (hypothalamic-pituitary-adrenal axis), congestive heart failure, hypertension, emotional changes, thinning of skin, petechiae, diabetes mellitus, menstrual irregularities, electrolyte imbalances, liver and pancreatic dysfunction, exophthalmos, glaucoma, loss of muscle mass, and muscle weakness. Because corticosteroids affect the HPA system, gradual withdrawal by tapering the dose over time is necessary when discontinuing long-term therapy.
Cytotoxins, such as methotrexate, are used to manage autoimmune disorders because of their ability to kill actively proliferating lymphocytes after they are transformed from their resting G0 state. The key to the use of cytotoxins is to effectively apply their killing activity without damaging the rest of the body. Cyclosporine (Sandimmune) is a more selective immunosuppressant that reversibly suppresses T-helper cells in the G0 or G1 phase of the cell cycle without killing them. As a result, it inhibits the development of killer or cytotoxic T cells without decreasing the numbers of cells. It also impairs the ability of T cells to respond effectively to foreign antigens. It is used to suppress reactions during tissue or organ transplantation. Frequent side effects include edema, hypertension, headache, hirsutism, elevated triglyceride levels, gastrointestinal effects, nephropathy, infection, emotional changes, gynecomastia, leukopenia, anemia, and hepatotoxicity.
Tumor necrosis factor (TNF) inhibitors or immunomodulators, such as etanercept (Enbrel) or infliximab (Remicade), are used as disease modifiers that bind to and block the activity of TNF-α and TNF-β. They may also modulate TNF-mediated responses, such as leukocyte migration and expression of adhesion molecules. The most common side effects of TNF inhibitors include headache, gastrointestinal changes, injection site skin reactions, respiratory tract infections, edema, dizziness, dyspepsia, and weakness.
Therapeutic plasmapheresis is another type of therapy occasionally used in the management of autoimmune diseases. Plasmapheresis is analogous to dialysis and involves the selective filtering or removal of plasma or a plasma cell type as well as protein-bound toxic substances. The patient’s whole blood is filtered, blood cells and platelets are returned, and the plasma component containing the autoantibodies is removed and replaced with 5% albumin or another colloid solution. According to the American Society for Apheresis, this type of therapy has been effective in the management of diseases such as myasthenia gravis, thrombocytopenia purpura, multiple sclerosis, and Rh-negative hemolytic disease of the newborn. Plasmapheresis is generally well tolerated; however, there are both major and minor risks involved in the process. Some of the risks include insertion of large intravenous (IV) catheters to perform the procedure, decrease in serum ionized calcium concentration, shifts of fluid levels, and risks of infection and bleeding from loss of coagulation factors.
Hypersensitivity
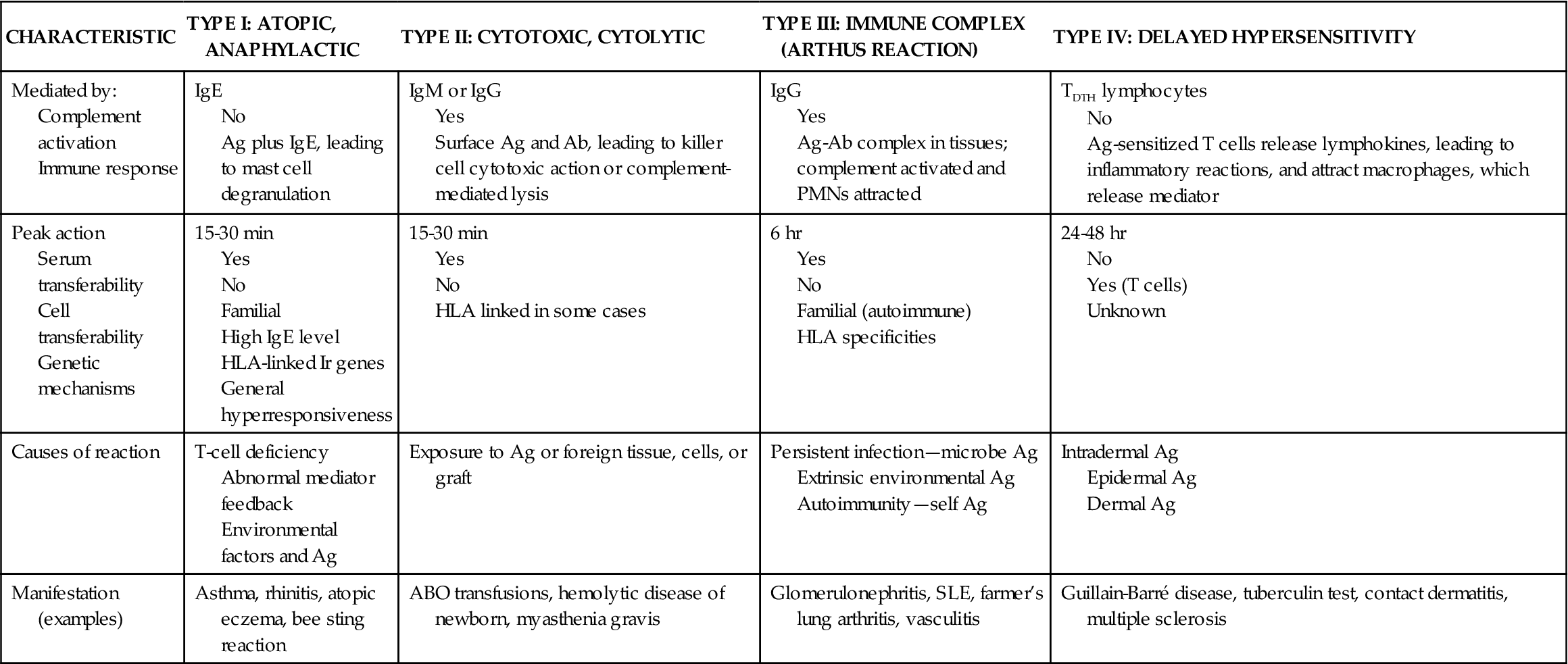
Hypersensitivity is a normal immune response that is inappropriately triggered or excessive or produces undesirable effects on the body. The basic mechanism that triggers hypersensitivity is a specific antigen-antibody reaction or a specific antigen-lymphocyte interaction. Four classes or types of hypersensitivity are differentiated. Each type is characterized by a specific cellular or antibody response. Hypersensitivity types I, II, and III are mediated by antibodies produced by B lymphocytes. Type IV hypersensitivity is mediated by T cells. Hypersensitivity reactions are specific to a particular antigen and usually do not occur on first exposure to the antigen. Although the diseases or syndromes associated with each type differ in their clinical signs and symptoms, the underlying pathophysiologic process is similar within each type. In Table 10-2 the four major types of hypersensitivity are contrasted. The complex interactions between immune system inflammatory mediators, cytokines, T cells, B cells, and mast cells characterize hypersensitivity reactions.
TABLE 10-2
THE FOUR TYPES OF HYPERSENSITIVITY
| CHARACTERISTIC | TYPE I: ATOPIC, ANAPHYLACTIC | TYPE II: CYTOTOXIC, CYTOLYTIC | TYPE III: IMMUNE COMPLEX (ARTHUS REACTION) | TYPE IV: DELAYED HYPERSENSITIVITY |
| Mediated by: Complement activation Immune response | IgE No Ag plus IgE, leading to mast cell degranulation | IgM or IgG Yes Surface Ag and Ab, leading to killer cell cytotoxic action or complement-mediated lysis | IgG Yes Ag-Ab complex in tissues; complement activated and PMNs attracted | TDTH lymphocytes No Ag-sensitized T cells release lymphokines, leading to inflammatory reactions, and attract macrophages, which release mediator |
| Peak action Serum transferability Cell transferability Genetic mechanisms | 15-30 min Yes No Familial High IgE level HLA-linked Ir genes General hyperresponsiveness | 15-30 min Yes No HLA linked in some cases | 6 hr Yes No Familial (autoimmune) HLA specificities | 24-48 hr No Yes (T cells) Unknown |
| Causes of reaction | T-cell deficiency Abnormal mediator feedback Environmental factors and Ag | Exposure to Ag or foreign tissue, cells, or graft | Persistent infection—microbe Ag Extrinsic environmental Ag Autoimmunity—self Ag | Intradermal Ag Epidermal Ag Dermal Ag |
| Manifestation (examples) | Asthma, rhinitis, atopic eczema, bee sting reaction | ABO transfusions, hemolytic disease of newborn, myasthenia gravis | Glomerulonephritis, SLE, farmer’s lung arthritis, vasculitis | Guillain-Barré disease, tuberculin test, contact dermatitis, multiple sclerosis |
Type I Hypersensitivity
Etiology
Genetic mechanisms influence type I hypersensitivity with strong genetic or hereditary linkage regarding the IgE response to antigens (allergens). This genetic component involves both the ability to respond to an allergen and the general ability to produce an IgE antibody response. For example, children born to two allergic parents have a 50% chance of being allergic. Children born to one nonallergic and one allergic parent have a 30% chance of being allergic. It has been identified that total IgE concentration is higher in patients with atopic rhinitis or asthma compared to nonatopic patients.8
Pathogenesis
Type I hypersensitivity is also known as immediate hypersensitivity, because the reaction is immediate. It is a sensitization reaction characterized by signs and symptoms of an allergic reaction that usually occurs 15 to 30 minutes after exposure to an antigen (allergen).
At the cellular level, immunoglobulin E (IgE) is the principal antibody mediating this reaction. IgE is produced by specialized plasma B cells and circulates in very small amounts in the blood. When an individual is exposed to an allergen, selected plasma B cells produce allergen-specific IgE. It usually takes repeated exposures to the allergen to cause significant levels of IgE to be present in the blood. Environmental pollutants may play a role by increasing mucosal permeability and enhancing antigen (allergen) entry into the body. This increased entry would subsequently increase IgE responsiveness.
Mast cells and basophils are the principal effector cells, although there are many other cells with histamine and other inflammatory mediators that can be involved in the reaction. These may include neutrophils, eosinophils, lymphocytes, macrophages, epithelial cells, and endothelial cells. Mast cells are found throughout the body in all loose connective tissue. They are covered with IgE receptors—up to 500,000 on their cell surfaces—and they are filled with vesicles or granules containing potent vasoactive, proinflammatory chemical mediators (especially histamine) that produce inflammation when they are released. The IgE receptors on mast cells bind the Fc portion of an IgE antibody. The IgE antigen-binding sites are then displayed on the mast cell surface, where they can bind to antigens that pass by the mast cell (Figure 10-1). This process makes the mast cells responsive to particular antigens.
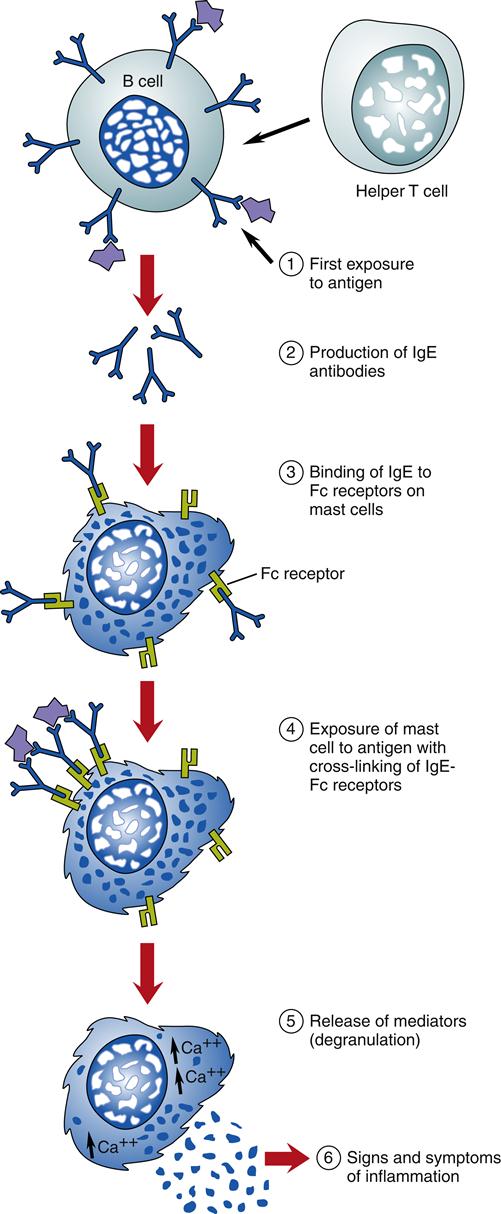
The initial incident during a type I hypersensitivity response is the cross-linking of two IgE receptors to one antigen on the mast cell located at the site of the allergen’s entry into the body (see Figure 10-1). Cross-linking of IgE and the antigen causes an increase in intracellular calcium (Ca2+) concentration that results in immediate, massive, local mast cell degranulation of preformed and newly formed proinflammatory mediators. The release of mediators causes an inflammatory response.
Mast cells, basophils, and other effector cells release many chemicals. Some of the mediators are preformed and stored in vesicles, such as histamine, heparin, proteolytic enzymes, and chemotactic factors. Other mediators are formed during the degranulation process. Examples of newly formed mediators include superoxide, prostaglandins, thromboxanes, leukotrienes, bradykinin, and interleukins (see Chapter 9).
One of the most important mediators of type I hypersensitivity is histamine. Histamine binds to H1 (histamine 1), H2, H3, and H4 receptors, which are located on many types of cells. Mast cells have receptors for H1, H2, and H3, with H1 receptors being the most active. Basophils express predominantly H2 receptors, whereas neutrophils and eosinophils have both H1 and H2 receptors. Recent evidence shows that H1 and H2 receptors are present on monocytes and macrophages, with an increase in H1 receptors when monocytes differentiate into macrophages. Histamine binding to H1 receptors triggers increased vascular permeability, vasodilation (flushing), urticaria formation (hives), smooth muscle constriction (bronchoconstriction), increased mucus secretion and pruritus (increased itching), and increased gut permeability. The activation of H2 receptors has opposing effects to H1 receptors in some tissues and causes smooth muscle relaxation in the lower airways, augments gastric acid secretion from parietal cells, and in high concentrations has an inhibitory effect on inflammatory cells, decreasing degranulation and decreasing neutrophil chemotaxis. The H3 receptors are located in the brain, in the spinal cord, and on sensory neurons such as postganglionic cholinergic nerves in lung bronchi. The H4 receptors are found on immune system cells such as dendritic cells, eosinophils, T cells, monocytes, macrophages, and natural killer cells as well as in hematopoietic cells such as the spleen, thymus, bone marrow, and blood leukocytes. H4 receptors are highly attracted to histamine and are also involved in chemotaxis and inflammatory responses.
The proteolytic enzymes kininogenase and tryptase activate the kinin pathway and C3 activates the complement cascade via the alternative pathway. Heparin decreases clot formation. The chemotactic factors recruit or activate other inflammatory and immune cells. Leukotrienes cause smooth muscle contraction and increase vascular permeability.
Clinical manifestations
Manifestations of an immediate hypersensitivity reaction vary in severity and intensity. For many people, type I hypersensitivity reactions are annoying, such as hives (urticaria), seasonal allergic rhinitis, eczema, or mild bronchoconstriction. In other people, the symptoms are more problematic, including tightening of the throat, localized edema, wheezing, and tachycardia, such as is associated with localized angioedema reactions or severe airway reactions. In a very small number of highly allergic people, the type I hypersensitivity reaction can be expressed as a life-threatening allergic reaction known as anaphylaxis such as that associated with bee stings and seafood or peanut allergic reactions. Common allergenic medications, insects, and foods that can trigger type I hypersensitivity reactions are listed in Box 10-1.
Treatment
Treatment for type I hypersensitivity primarily involves pharmacologic management with antihistamines, β-adrenergics, corticosteroids, anticholinergics, and anti–immunoglobulin E therapy (IgE blocker therapy). Antihistamines such as diphenhydramine (Benadryl) are used to block the effect of histamine. This action decreases vascular permeability and bronchoconstriction. β-Adrenergic sympathomimetics are used to decrease bronchoconstriction and bronchospasm. Epinephrine is an adrenergic agent (α, β1, and β2) given subcutaneously or intravenously during acute allergic reactions, especially after food or bee sting reactions. Most patients with severe allergies to food or insect bites are given prescriptions for epinephrine in the form of EpiPen with an autoinjector. Corticosteroids are used to decrease the inflammatory response. Anticholinergics are used to block the parasympathetic system and thus allow greater sympathetic activity. This action indirectly causes bronchodilation. Anti–immunoglobulin E therapy (omalizumab) may be used for persons with severe persistent asthma. Omalizumab (Xolair) is a subcutaneously injected, monoclonal anti-IgE antibody that binds to the IgE molecule, thus inhibiting the binding of IgE to mast cells and basophils and promoting down-regulation of IgE receptors. It is used to improve asthma control in patients with moderate to severe persistent asthma not controlled with inhaled corticosteroids. It is used in children 12 years or older and in adults.
Prevention
Some protective, proactive actions taken during pregnancy are thought to decrease the likelihood that type I hypersensitivity will develop in children from families with a history of allergies. These actions include avoiding foods to which the mother is allergic, limiting excesses of one type of food during the last trimester of pregnancy, avoiding whole eggs during the last month before delivery and while breast feeding, and limiting cow’s milk to two glasses per day. Other actions that may be helpful during the child’s infancy include avoiding exposure to environmental pollution, breast feeding for a minimum of 6 months, supplementing the child’s diet with non–cow’s milk products such as soy milk, giving solid foods only after the infant is 6 months old, keeping the infant’s room as free of dust and molds as possible, and keeping pets (dogs, cats, birds) out of the home.
Pharmacotherapeutic prevention
Another avenue for prevention of type I hypersensitivity reactions involves the use of desensitization therapy (immunotherapy). Desensitization, or immunotherapy, is more successful in patients with hay fever than in those with other types of allergies. It involves both environmental control of external allergens and titrated pharmacologic exposure to allergens. Environmental control involves a systematic plan to decrease exposure to house dust, molds, and animal dander. Pets are kept out of the house. The person must avoid food allergens, wool carpets, goose down or feather pillows, dried plants, and exposure to other animal and vegetable products. The person is urged to use air conditioning and electronic air filters.
Pharmacologic desensitization involves injecting a person with sufficient antigen (allergen) on a regular basis over a course of months or years, followed by periodic maintenance or booster therapy. Gradually the dose is increased until the person can tolerate the allergen without a type I hypersensitivity reaction. The goal of this therapy is a change in immunoglobulins so that there is an increase in IgG- and IgA-blocking antibodies, no increase in IgE during allergy season, decreased basophil reactivity, and decreased lymphocyte reactivity to allergens.
Type II Hypersensitivity
Etiology and pathogenesis
Type II hypersensitivity, also known as tissue-specific, cytotoxic, or cytolytic hypersensitivity, is characterized by antibodies that attack antigens on the surface of specific cells or tissues. Often the reaction is immediate (15 to 30 minutes after exposure to the antigen). However, it can occur over time, such as in thyroiditis or myasthenia gravis. The mechanisms that encompass type II tissue-specific hypersensitivity all occur after the binding of antibody to tissue-specific antigens. The reaction is mediated by the complement system and a variety of effector cells, including tissue macrophages, platelets, natural killer cells, neutrophils, and eosinophils. IgG and IgM are the principal antibodies. Examples of this type of hypersensitivity reaction include ABO transfusion reactions, hemolytic disease of the newborn, myasthenia gravis, thyroiditis, hyperacute graft rejection, and autoimmune hemolytic anemia (Table 10-3). Transfusion reactions, hemolytic disease of the newborn, and graft rejection are examples of isoimmunity (alloimmunity), a condition in which the immune system reacts against antigens on tissues from other members of the same species.
TABLE 10-3
DISEASE AND AUTOANTIBODIES ASSOCIATED WITH TYPE II HYPERSENSITIVITY
| DISEASE | ANTIGEN/AUTOANTIBODY |
| Type 1 diabetes | Islet cells |
| Insulin-resistant diabetic states | Insulin receptor |
| Myasthenia gravis | Acetylcholine receptor |
| Addison disease | Adrenal epithelial cells |
| Autoimmune hemolytic anemia | Red blood cell membrane |
| Immune thrombocytopenic purpura | Platelet membrane |
| Autoimmune neutropenia | Neutrophil antigens |
| Pernicious anemia | Intrinsic factor, gastric parietal cells |
| Lymphocytic thyroiditis | Thyroglobulin |
| Graves disease | Receptor for thyroid-stimulating hormone |
| Pemphigus vulgaris | Desmosomes |
| Hyperacute graft rejection | Donor antigens |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


