Alterations in Cardiac Function
Shann D. Kim and Jacquelyn L. Banasik
Key Questions
• What factors alter the balance between myocardial oxygen supply and demand?
• How do the clinical features of the coronary heart disease syndromes differ?
• How do valvular disorders alter cardiac pressure dynamics and workload?
• What are the similarities and differences among the cardiomyopathies and myocarditis?
• How do pericarditis and pericardial effusions differ in regard to cause and significance?
• What factors determine whether a congenital heart defect will produce cyanosis?

http://evolve.elsevier.com/Copstead/
The incidence of cardiovascular disease (CVD) increased rapidly in the United States during the last century, but the death rates from CVD declined between 1998 and 2008 by 30.6%.1 Mortality data from 2007 showed that CVD accounted for 32.8% of all deaths. Approximately half of these deaths are due to coronary heart disease (CHD), whereas stroke, high blood pressure, heart failure, and others claim the remainder. Since the late 1960s, however, a decline in cardiac mortality has been achieved in the United States because of improvements in treatment and prevention. More than 16 million people living today have a history of angina pectoris or myocardial infarction (MI).1 Men and women are equally represented, although women tend to be older when their heart disease becomes apparent. In 2008, the direct and indirect economic cost of cardiovascular diseases, including stroke, was estimated at $297.7 billion annually.1 CHD is the most important cardiovascular disorder in terms of numbers affected and economic impact.
Coronary Heart Disease
CHD is also called ischemic heart disease (IHD) and coronary artery disease (CAD) in some sources. These terms are related because CHD is characterized by insufficient delivery of oxygenated blood to the myocardium (ischemia) because of atherosclerotic coronary arteries (CAD). The American Heart Association compiles statistics under the heading of CHD, which includes the diagnoses of angina pectoris and myocardial infarction. CHD caused about one in six deaths in the United States in 2008.1 Other sequelae of CHD include dysrhythmias, sudden cardiac death, and heart failure. When metabolic demand for oxygen exceeds supply, the myocardium becomes ischemic, which leads to a dysfunction in cardiac pumping and predisposes to abnormal heart rhythms. If the ischemic episode is severe or prolonged, irreversible damage to myocardial cells may result in MI.
Etiology of Coronary Heart Disease
Atherosclerosis of coronary arteries is the source of nearly all CHD. Atherosclerosis causes progressive narrowing of the arterial lumen and predisposes to a number of processes that can precipitate myocardial ischemia, including thrombus formation, coronary vasospasm, and endothelial cell dysfunction. Uncommon causes of cardiac ischemia include abnormalities of blood oxygen content (e.g., respiratory failure) and poor perfusion pressure through the coronary arteries (e.g., hypotension, hypovolemia). Occasionally, patients experience the signs and symptoms of cardiac ischemia but show no evidence of significant coronary artery atherosclerosis when evaluated by angiography. These patients are thought to have abnormalities of the microcirculation. Abnormal vascular regulation by endothelial cells in small vessels of the heart has been suggested as a probable mechanism. Endothelial cells secrete variable quantities of vascular relaxing and contracting factors and play a key role in controlling myocardial blood flow. Abnormalities of the microcirculation are more difficult to detect than coronary artery plaque, which is evident on coronary angiography. As evaluation methods improve, disorders of the microcirculation are likely to be more frequently recognized as factors contributing to CHD.
Mechanisms of Coronary Atherosclerosis
Knowledge about mechanisms of plaque formation in the coronary arteries has rapidly accumulated in recent years. Epidemiologic studies reported in the 1960s suggested associations among certain traits and habits and the development of CHD. More recent studies have confirmed and expanded on these risk factors, which include several major risks (e.g., age, family history, abnormal lipid levels, cigarette smoking, hypertension, diabetes, and obesity) and numerous probable risks (Box 18-1).2 Although males and females succumb to heart disease in equal numbers, male gender is a risk factor for earlier development of heart disease, on average about 10 years earlier. The risk factors for CHD are the same as those for atherosclerosis in other arteries and are discussed in Chapter 15.
The observation that atherosclerotic plaque is composed primarily of lipid prompted the idea that abnormal lipid metabolism was a probable culprit in the development of CHD, and a great deal of attention has been focused on therapies to reduce levels of serum cholesterol in individuals with dyslipidemia. Lipids are transported through the bloodstream in combination with specific proteins (apoproteins). Certain lipid-protein molecules (lipoproteins) are associated with a greater risk of atherosclerosis. The five major kinds of lipoproteins are shown in Figure 18-1. High levels of low-density lipoproteins (LDLs), which are high in cholesterol, have been associated with the highest risk. Very-low-density lipoproteins, which have large amounts of triglycerides, also appear to increase the risk. High-density lipoproteins, on the other hand, have been correlated with a decreased risk of atherosclerosis.2
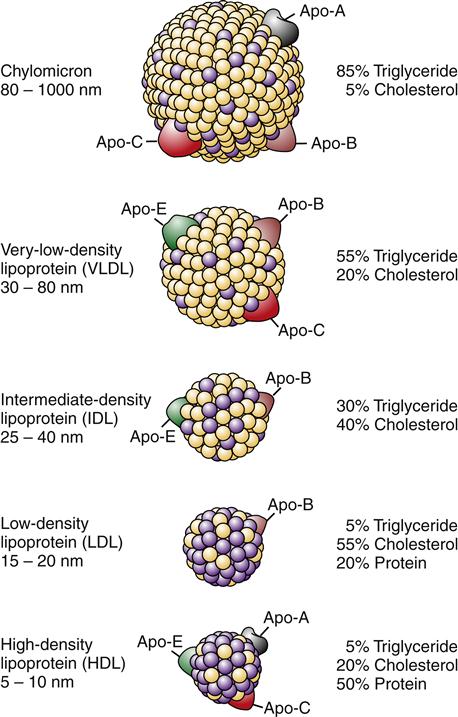
Binding of lipoproteins to receptors is mediated through apoproteins.
High-density lipoproteins are thought to transport cholesterol from the peripheral tissues back to the liver, thus removing atheromatous plaque. The role of low-density and, indirectly, very-low-density lipoproteins is to transport cholesterol to the peripheral tissues (Figure 18-2). Cholesterol uptake by peripheral cells is mediated by LDL receptors on cell surfaces that bind and promote endocytosis of cholesterol. The liver normally binds and internalizes about 75% of the circulating LDL cholesterol.
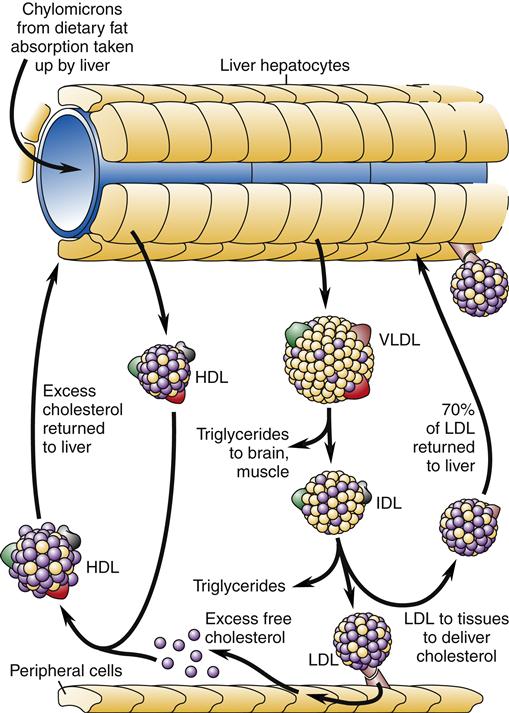
Chylomicrons from dietary fat absorption are taken up by the liver and resynthesized into high-density lipoprotein (HDL) and very-low-density lipoprotein (VLDL). HDL circulates to the peripheral tissues and takes up excess cholesterol for transport back to the liver. Triglycerides are removed for tissue use from VLDL, which becomes intermediate-density lipoprotein (IDL). More triglyceride removal leads to the formation of low-density lipoprotein (LDL). LDL is absorbed by peripheral tissues to obtain cholesterol. About 70% of the circulating LDL returns to the liver.
Extreme cases of hyperlipidemia occur in individuals who have genetic derangements in lipid metabolism. These disorders run in families, and some are associated with the development of severe coronary atherosclerosis at a young age unless aggressively managed. The most common form of genetic hyperlipidemia (familial hypercholesterolemia) is associated with a defect in the LDL receptor on liver cells.3 Inability of the liver to efficiently remove cholesterol from the bloodstream results in hyperlipidemia. Genetic disorders of lipid metabolism are described in Table 18-1. Even when lipid metabolism is normal, a high-fat diet can overwhelm the liver’s ability to clear LDL cholesterol from the circulation and results in hyperlipidemia. Dietary fat restriction may be beneficial in reducing cholesterol level in this case.
TABLE 18-1
| DISORDER | GENE |
| LDL Particles | |
| Familial hypercholesterolemia | LDL-R |
| Familial defective ApoB-100 | ApoB |
| Autosomal dominant hypercholesterolemia | PCSK9 |
| Autosomal recessive hypercholesterolemia | ARH |
| Abetalipoproteinemia | MTP |
| Hypobetalipoproteinemia | ApoB |
| Familial sitosterolemia | ABCG5/ABCG8 |
| Familial LP(a) hyperlipoproteinemia | Apo(a) |
| Remnant Lipoproteins | |
| Dysbetalipoproteinemia type III | ApoE |
| Hepatic lipase deficiency | HL |
| Triglyceride-Rich Lipoproteins | |
| Lipoprotein lipase deficiency | LPL |
| ApoC-II deficiency | ApoC-II |
| Apo-AV | ApoA-V |
| Familial hypertriglyceridemia | Polygenic |
| Familial combined hyperlipidemia | Polygenic |
| HDL Particles | |
| Apo-A1 deficiency | Apo-A1 |
| Tangier disease, familial HDL deficiency | ABCA1 |
| Familial LCAT deficiency syndromes | LCAT |
| CETP deficiency | CETP |
| Niemann-Pick disease types A and B | SMPD1 |
| Niemann-Pick disease type C | NPC1 |
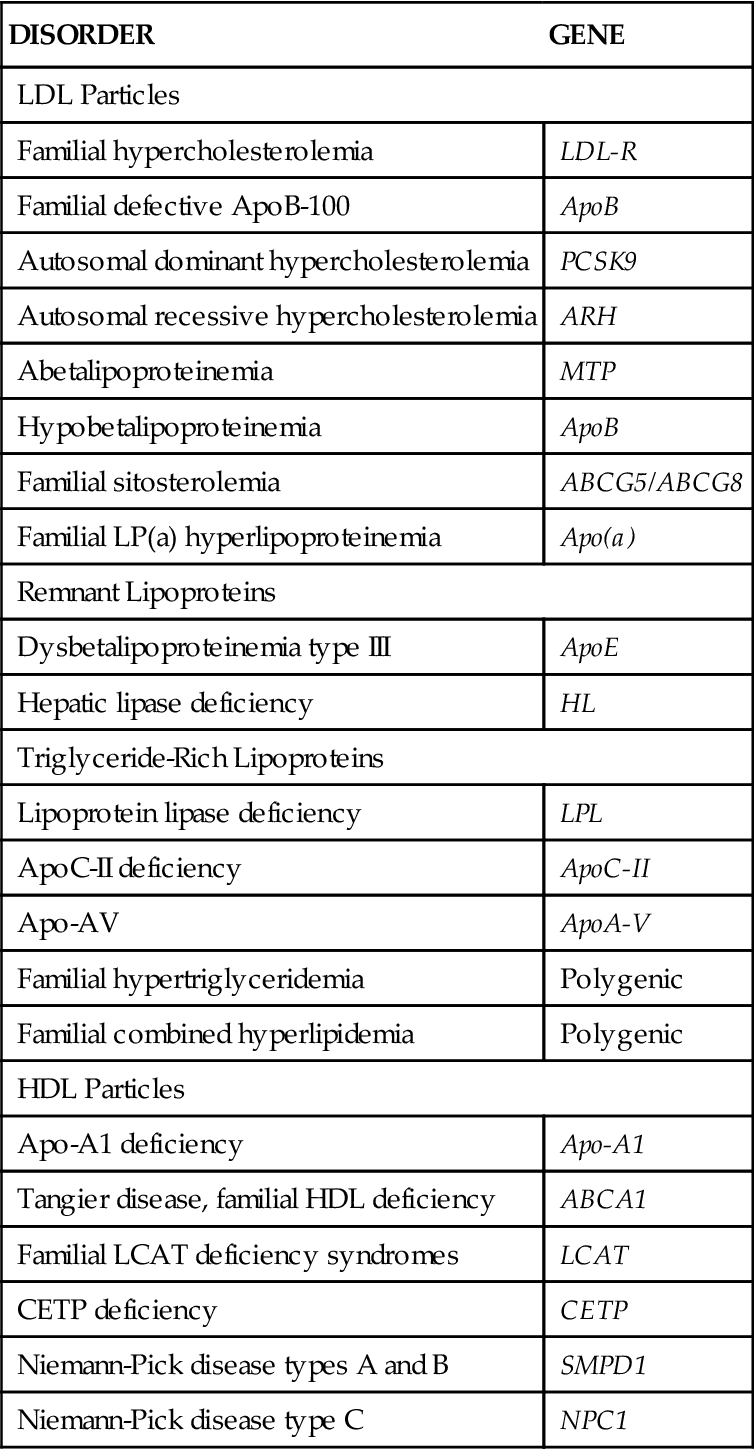
Adapted from Genest J, Libby P: Lipoprotein disorders and cardiovascular disease. In Bonow R, Mann D, Zipes D et al, editors: Braunwald’s heart disease: a textbook of cardiovascular medicine, ed 9, Philadelphia, 2012, Saunders, p 983.
Atherosclerotic plaque formation is initiated by injury to the coronary artery endothelium. The specific cause of endothelial dysfunction in the early stage of atherosclerosis is uncertain; however, several potential mechanisms have been described. These include chronic hemodynamic wall stress, which may explain the typical localization of plaques at arterial branch points and the role of hypertension as a risk factor; toxins from cigarette smoke; circulating inflammatory cytokines; and hyperlipidemia. Once the injury occurs, the endothelium may become more permeable and recruit leukocytes (Figure 18-3). LDLs leak through the endothelium and into the vessel wall (insudation) where they are oxidized by endothelial cells and macrophages.4 Oxidized lipids are damaging to the endothelial and smooth muscle cells, and stimulate the recruitment of macrophages into the vessel wall, where they engulf the lipids. Lipid-filled macrophages are called foam cells. The macrophages and foam cells release inflammatory mediators and growth factors that attract more leukocytes and stimulate smooth muscle proliferation. Excess lipid and debris begins to accumulate within the vessel wall and to coalesce into a pool called the lipid core (Figure 18-4). Atherosclerotic plaques with large lipid cores are fragile and prone to rupture. Rupture of a plaque exposes subendothelial proteins and initiates platelet aggregation and thrombus formation. Thrombi may be asymptomatic if they are small and do not occlude the artery. Components of the thrombus may be incorporated into the plaque, causing it to enlarge. Older plaques have significant collagen and fibrin, which form a cap and tend to make the plaque more stable. Numerous therapies aimed at stabilizing vulnerable plaques and preventing thrombus formation have been studied in clinical trials. Lipid-lowering therapy is a mainstay of treatment and prevention for atherosclerosis. Targets for serum LDL cholesterol levels have been developed by the National Cholesterol Education Program (NCEP) Adult Treatment Panel (NCEP III) based on the presence of known CHD or risk factors (Table 18-2).5 An update to the NCEP III is expected in 2012. In addition to risk factor modification, therapies to reduce plaque inflammation, inhibit lipid oxidation, and prevent thrombosis are in common use (Table 18-3). A major aim of therapy is to stabilize the plaques, making them less prone to rupture.
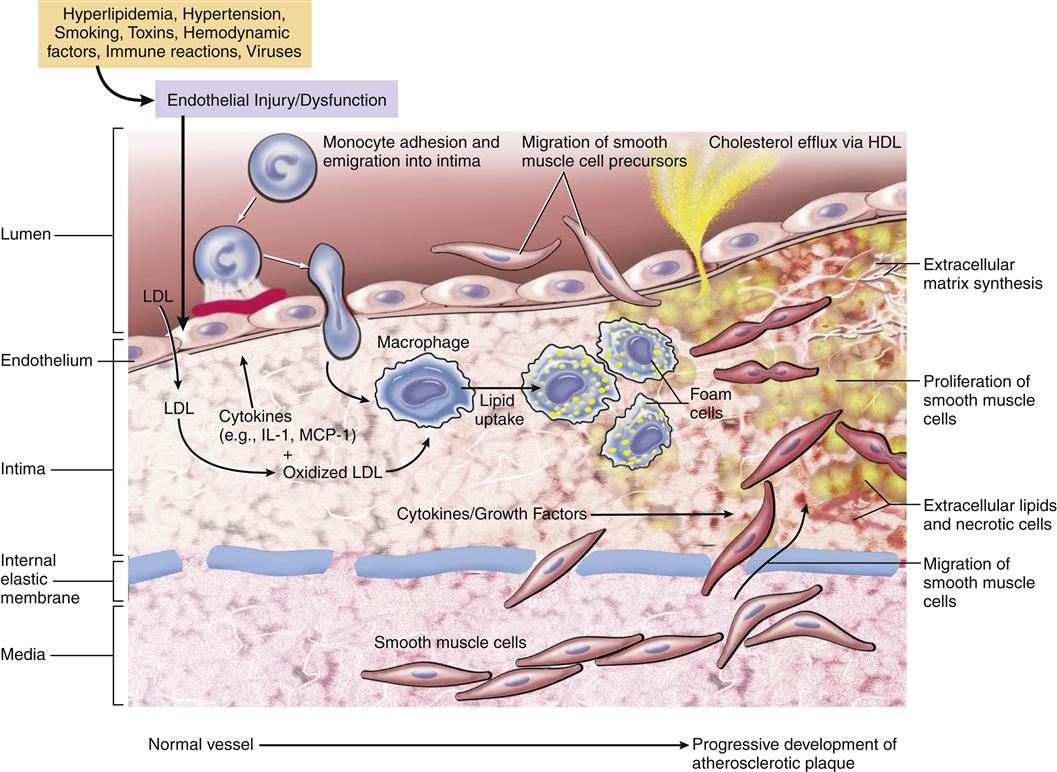
Note that smooth muscle cells migrate from the intima through the internal elastic membrane and into the intimal layer where they proliferate in response to growth factors. Macrophages in the intima release signals that alter the endothelial cell layer and induce expression of cell adhesion molecules that recruit monocytes into the tissue. (From Kumar V et al, editors: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2010, Saunders, p 501.)
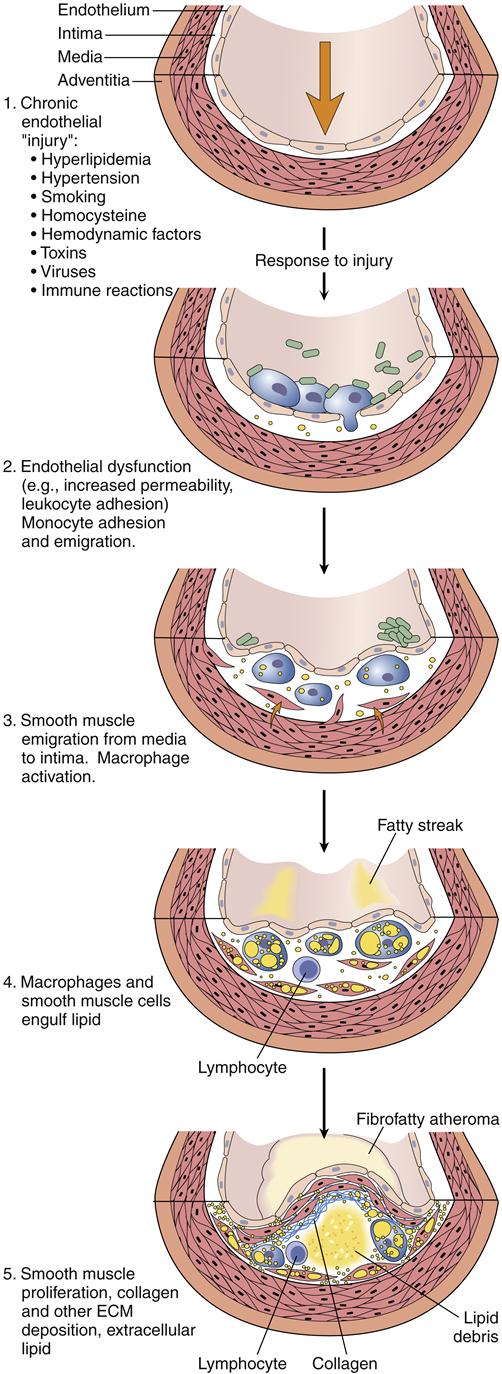
1, Chronic endothelial injury leads to 2. 2, Endothelial dysfunction, permeability, and inflammation. 3, Activated monocytes infiltrate the arterial wall and smooth muscle proliferates. 4, Macrophages engulf lipid to become foam cells. 5, A lipid core forms in the arterial wall and a fibrous cap evolves. (From Kumar V et al, editors: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2010, Saunders, p 499.)
TABLE 18-2
RECOMMENDED SERUM LOW-DENSITY LIPOPROTEIN TARGETS TO REDUCE THE RISK OF CORONARY HEART DISEASE
| PATIENT RISK CATEGORY | LDL-C CUT POINT FOR INITIATING DRUG THERAPY (mg/dl) | LDL-C GOAL (mg/dl) |
| High risk: CHD present or CHD risk equivalent (≥2 risks plus 10-year risk >20%) | >100 | <100 (optional <70) |
| Moderately high risk: ≥2 risks and 10-year risk 10-20% | ≥130 | <130 |
| Moderate risk: ≥2 risks and 10-year risk <10% | ≥160 | <130 |
| Lower risk: ≤1 risk | ≥190 | <160 |
CHD, Coronary heart disease; LDL-C, low-density lipoprotein cholesterol.
From National Cholesterol Education Program: Third Report of the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III), Circulation 110:227-239, 2004.
TABLE 18-3
ACTIONS OF THERAPIES TO REDUCE CORONARY HEART DISEASE
| THERAPY | MAJOR ACTIONS | OTHER ACTIONS |
| Angiotensin inhibitors (ACEI, ARB) | Improve endothelial function | Antioxidant (LDL), antiinflammatory |
| Statins | Decrease LDL-C, increase HDL-C | Improve endothelial function, antiinflammatory, antioxidant (LDL) |
| Fish oil (omega-3) | Decreases LDL-C, increases HDL-C | Inhibits thrombosis |
| Fibrates | Improve endothelial function | Increase HDL |
| Aspirin | Inhibits thrombosis, antiinflammatory | |
| Exercise | Increases HDL | Improves endothelial function |
Atherosclerotic lesions generally increase in size over many years and progressively occlude the lumen of vessels. A significant reduction in blood flow can result when plaque occupies 75% or more of the arterial lumen. Clinically significant atherosclerotic plaque may be located anywhere within the three major coronary arteries or major secondary branches. All three coronary arteries are often simultaneously affected, although some individuals have only one or two diseased vessels. Surprisingly, the extent and severity of atherosclerotic lesions are not good predictors of the severity of ischemia.
Atherosclerotic coronary lesions have been characterized and attempts made to correlate the anatomic descriptions with plaque development and behavior. Typically atherosclerotic lesions begin as fatty streaks and progress to small regions of medial wall thickening with scattered macrophages at a young age. These lesions are considered to be precursor lesions and are not symptomatic.6 As the plaques acquire more free lipid within the arterial wall, they are more vulnerable to rupture, thrombus formation, and progressive plaque growth. These are considered to be advanced lesions and carry a significant risk of producing disruptions in coronary blood flow. Critical narrowing of the coronary lumen over time or sudden rupture of a plaque followed by thrombus formation causes the clinical syndromes of CHD, including angina, infarction, ischemic cardiomyopathy, and sudden cardiac arrest.
Stable plaques usually are asymptomatic or may be associated with exercise-induced angina pain (stable angina pectoris). However, plaques are vulnerable to rupture or erosion, which can initiate thrombus formation and acute coronary occlusion. A variety of factors have been identified as markers of increased plaque vulnerability. These factors include (1) active inflammation within the plaque; (2) a large lipid core with a thin cap; (3) endothelial denudation (erosion) with superficial platelet adherence; (4) fissured or ruptured cap; and (5) severe stenosis predisposing to high shear stress.7 Acute coronary syndrome (ACS), or unstable angina or MI, as well as sudden cardiac arrest, is nearly always associated with acute disruption of a vulnerable plaque. Because the types of plaques that are most vulnerable often do not significantly obstruct the lumen before they rupture, ACS frequently occurs in individuals whose disease had been asymptomatic. Patients with a high risk for or known presence of vulnerable plaques benefit from therapies such as lipid-lowering agents (to stabilize plaques) and antiplatelet agents (to prevent thrombosis).8
Pathophysiology of Ischemia
Ischemia of cardiac cells occurs when the oxygen supply is insufficient to meet metabolic demands. Myocardial cells are unable to store much energy in the form of adenosine triphosphate (ATP) and must therefore continuously receive a supply of oxygen for aerobic synthesis of ATP. ATP is essential for powering myocardial contraction as well as for cell maintenance. Because the heart is unable to slow its activity when ATP supplies dwindle, a steady flow of oxygen is essential.
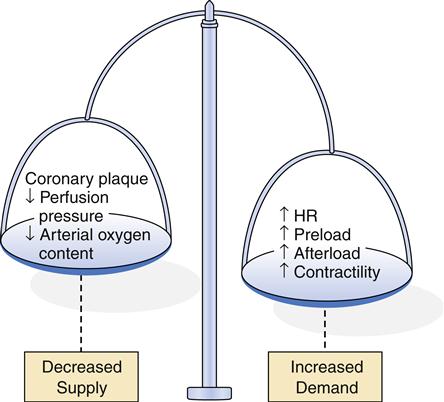
Factors that decrease myocardial oxygen supply or increase myocardial oxygen demand can upset the balance and result in cellular ischemia. Thus, the critical factors in meeting cellular demands for oxygen are (1) the rate of coronary perfusion and (2) the myocardial workload. Coronary perfusion can be impaired in several ways, including (1) large, stable atherosclerotic plaque, (2) acute platelet aggregation and thrombosis, (3) vasospasm, (4) failure of autoregulation by the microcirculation, and (5) poor perfusion pressure.
Myocardial workload depends on heart rate, preload, afterload, and contractility (see Chapter 17). An increase in any of these variables increases myocardial oxygen requirements and may precipitate ischemia. However, even conditions resulting in very high myocardial oxygen consumption will seldom lead to ischemia unless some underlying impairment in coronary perfusion is present.
One or more of the aforementioned mechanisms are operative in producing clinically significant myocardial ischemia resulting in the acute or chronic coronary syndromes. Advanced fibrous plaque is thought to produce intermittent ischemia when 75% or more of the arterial lumen is occluded.9 Because fibrous plaque progresses slowly over many years, the heart can develop alternative pathways for myocardial blood flow. This collateral circulation can preserve blood flow despite almost total occlusion of the coronary artery. Thus, stable fibrous plaque may produce no symptoms of ischemia unless the demand of the heart for oxygen is suddenly elevated, as occurs in exercise or stress. When the onset of ischemia is predictable with certain activities and subsides with rest, the patient is said to have a chronic coronary syndrome, called classic or stable angina pectoris.
ACS occurs when sudden obstruction of coronary blood flow results in acute myocardial ischemia. Acute obstruction is usually associated with the formation of a clot in the coronary artery at the site of a vulnerable plaque. Rupture of the plaque exposes a rough area composed of collagen and other molecules that are thrombogenic. A high fibrinogen level, as occurs in smokers, and enhanced platelet adhesiveness, as occurs in hyperlipidemia, may enhance the risk of thrombus formation. Clot formation begins with adherence of platelets to the ruptured plaque. The platelets that initially attach release chemicals that attract more platelets, which aggregate and form a plug. The coagulation cascade may also be initiated and result in the formation of a platelet-fibrin clot that may occlude the vessel or break loose and travel farther along the vessel. Chemicals released by activated platelets include several vasoactive products (e.g., serotonin, thromboxane) that may contribute to spasm of the coronary vessel, further reducing blood flow.
Thrombosis occurs suddenly and may partially or completely obstruct the artery and cause acute ischemia. The ACS may present as unstable angina, MI, or sudden cardiac arrest. Appreciation of the role thrombus formation plays in coronary obstruction has resulted in the prophylactic use of antithrombotics, such as aspirin. Research indicates that the long-term use of small doses of aspirin reduces mortality from ischemic heart disease.10
Vasospasm usually occurs in areas of atherosclerotic plaque, but is also proposed as a mechanism of ischemia in patients who have anginal signs and symptoms but no significant amount of fibrous plaque in the coronary arteries. Variant, or Prinzmetal, angina is the term applied to vasospasm-initiated anginal symptoms. The etiology of spasm in vessels having no significant atherosclerotic plaques is unclear, but usually responds promptly to vasodilating agents. Intense vasospasm can occur in response to certain drugs, such as cocaine.
As previously mentioned, endothelial cells are important regulators of vascular tone. They secrete variable amounts of constricting and relaxing factors to control tissue perfusion. This autoregulation of blood flow allows the microvasculature to dilate when the need for oxygen in a particular area is increased. Failure of endothelial cells to appropriately regulate flow is a potential mechanism of myocardial ischemia.11 Endothelial cells can be damaged by circulating toxins from cigarette smoke, immune cells, and infectious agents. Inflammatory disorders that may alter endothelial cell function include lupus erythematosus, Kawasaki syndrome, and polyarteritis nodosa. The importance of inflammatory processes in the pathogenesis of CHD has been recognized, resulting in efforts to find markers (e.g., serum high-sensitivity C-reactive protein) and methods to reduce inflammation in those at risk.
Even if the coronary arteries and microcirculation are functioning properly, coronary perfusion may still be inadequate if perfusion pressure is low. Recall from Chapter 17 that coronary blood flow occurs primarily during diastole and depends on the driving pressure in the aorta. A fall in aortic blood pressure can significantly reduce coronary perfusion, particularly in vessels with high resistance to flow. Conditions such as shock, hemorrhage, and anesthesia may be associated with a decline in blood pressure, which decreases driving pressure and coronary perfusion and results in myocardial ischemia. However, the most common cause of cardiac ischemia is atherosclerotic coronary arteries.
Clinical Features and Management of Coronary Syndromes
Five syndromes can be differentiated according to the severity and onset of cardiac symptoms. Stable angina pectoris and ischemic cardiomyopathy are chronic syndromes that usually progress slowly and are a consequence of chronic obstruction from stable atherosclerotic plaques. ACS has an abrupt onset and life-threatening consequences and is associated with acute changes in plaque morphology and thrombosis. ACS includes unstable angina and MI. Unstable angina and MI are combined together because they are difficult to differentiate in the acute stage when therapeutic decisions must be made. Any of the coronary heart syndromes may precipitate sudden cardiac death and associated dysrhythmias.
Angina Pectoris
Angina pectoris literally means chest pain and is associated with intermittent myocardial ischemia. The length and the severity of the myocardial ischemia are insufficient to result in the acute death of cells. Bouts of chest pain and associated symptoms are generally recurrent and may be precipitated by conditions that increase myocardial oxygen demand, such as exercise, stress, sympathetic nervous system activation, and increased preload, afterload, heart rate, or muscle mass. Ischemic pain receptors from the myocardium travel to the central nervous system with the eighth cervical nerve and the first through fourth thoracic dorsal root ganglia. Sensory neurons from the jaw, neck, and arm also travel in these nerve trunks, so heart pain may be perceived as emanating from these body parts. This phenomenon is called referred pain. Anginal pain may be described as burning, crushing, squeezing, or choking. Pain is sometimes represented by expressions such as “an elephant is sitting on my chest” or by the patient placing a tight fist on the chest. Anginal pain may be mistakenly attributed to indigestion or dental pain. In some cases, patients have atypical symptoms of myocardial ischemia, such as back pain, fatigue, or weakness, rather than the classic symptom of chest pain.
Anginal ischemia, although temporary, may result in inefficient cardiac pumping with resultant pulmonary congestion and shortness of breath. Three patterns of angina pectoris have been described: (1) stable or typical angina, (2) Prinzmetal or variant angina, and (3) unstable or crescendo angina. All these patterns are associated with underlying coronary vessel disease and may be exhibited in a particular individual at different times and under different conditions. Unstable angina may progress to acute ischemia and is discussed in the Acute Coronary Syndrome section along with MI.
Stable angina
Stable angina is the most common form and is therefore called classic or typical angina. Stable angina is characterized by stenotic atherosclerotic coronary vessels that reduce coronary blood flow to a critical level. The stenosed arteries dilate poorly in response to increased myocardial oxygen requirements. Under conditions of increased myocardial workload, such as during physical exertion or emotional strain, coronary perfusion is inadequate and ischemia results (Figure 18-5). The onset of anginal pain is generally predictable and elicited by similar stimuli each time. Stable angina is generally relieved by rest and nitroglycerin, a drug that causes coronary and peripheral vasodilation, reduces preload, and, consequently, reduces myocardial workload.
Prinzmetal variant angina
Prinzmetal variant angina is characterized by unpredictable attacks of anginal pain. Although most individuals with Prinzmetal angina have significant coronary atherosclerosis, the onset of ischemic symptoms is unrelated to physical or emotional exertion, heart rate, or other obvious causes of increased myocardial oxygen demand. Vasospasm has been identified as the probable mechanism leading to variant angina, although the cause of the vasospasm is unknown. Proposed mechanisms include atherosclerosis-induced hypercontractility, abnormal secretion of vasospastic chemicals by local mast cells, and abnormal calcium flux across vascular smooth muscle. Variant angina responds well to treatment with calcium channel–blocking drugs, which inhibit vascular smooth muscle contraction.
Patients with angina are at risk for developing ACS and need aggressive treatment for risk factor reduction and therapies to reduce the risk of plaque rupture, thrombosis, and dysrhythmia.
Acute Coronary Syndrome
Unstable angina and MI are difficult to distinguish on the basis of clinical manifestations and are lumped together as ACS. Both are characterized by chest pain that may be more severe and lasts longer than the patient’s typical angina and may occur in individuals whose disease was previously asymptomatic. In both cases, plaque rupture with subsequent acute thrombus development is thought to occur. In unstable angina, the occlusion is partial or the clot is dissolved before the death of myocardial tissue. In MI, the occlusion is complete and the thrombus persists long enough for development of irreversible damage to myocardial cells resulting in necrosis. In the past, differentiation of unstable angina and MI was based on laboratory evaluation of serum biomarker levels (e.g., MB band of creatine kinase [CK-MB], troponins I and T). If cardiac biomarkers were elevated, which is indicative of necrosis, a diagnosis of MI was made; if not, a diagnosis of unstable angina was appropriate. In a time when monitoring and management of complications were the mainstay of treatment, this approach worked well. With the advent of reperfusion therapy, which is effective only if administered early in the course of infarction, the distinction between unstable angina and MI has become less clinically relevant. Because unstable angina and MI present a similar clinical picture in the acute phase, they have been combined in treatment protocols for ACS (Figure 18-6).12 Patients with chest pain and evidence of acute ischemia on the electrocardiogram (ECG) (ST-segment elevation [STEMI]) are candidates for acute reperfusion therapy. Patients presenting with symptoms of unstable angina and no ST elevation on the ECG may not benefit from reperfusion strategies, and antiplatelet drugs are a cornerstone of therapy.13 Differentiation between unstable angina and NSTEMI (non ST elevation MI) is made after obtaining cardiac necrosis markers; those patients with elevations are diagnosed with NSTEMI and those without elevations are diagnosed with unstable angina.
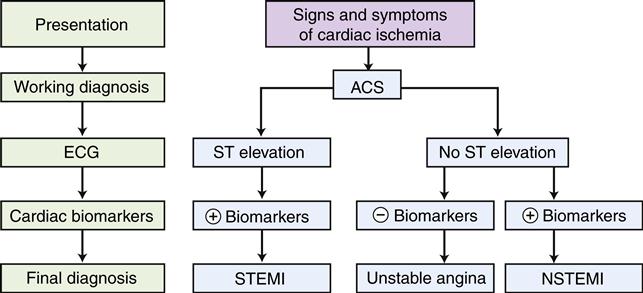
The electrocardiogram (ECG) is used to distinguish those patients with ST elevation from those with no ST elevation. Serum levels of cardiac biomarkers are then used to make a final diagnosis. Most patients with ACS characterized by ST elevation are diagnosed with MI (STEMI), and a proportion of patients with no ST elevation also will have elevated serum markers and are diagnosed with MI (NSTEMI). Patients who do not exhibit serum enzyme elevations are usually diagnosed with unstable angina.
Etiology and pathogenesis
MI results when prolonged or total disruption of blood flow to the myocardium causes cellular death by necrosis or apoptosis. Acute MI is an important form of CHD resulting in more than 150,000 deaths annually in the United States.1 It is estimated that an American male has a greater than 1 in 5 chance of sustaining an MI or fatal ischemic event before the age of 65. An MI may occur at any age, but the frequency rises with advancing age. Females younger than 45 years have a sixfold lesser risk of MI than men of the same age. After menopause, the rate of MI in women approaches that of their male counterparts and becomes essentially equal by age 80.9
As previously described, the initiating event in most MIs is believed to be development of a thrombus on top of an ulcerated or cracked atherosclerotic plaque. The initiating event is a sudden change in structure of the plaque. Platelets passing by the surface of the ruptured plaque adhere to it, initiate formation of a platelet plug, and activate the clotting cascade. The resultant thrombus grows until it occludes the vessel and triggers the transmural MI.
The thrombus theory of acute MI was controversial for many years because only 50% of persons dying of MI had a demonstrable thrombus at autopsy. Then DeWood and coworkers14 demonstrated that about 90% of persons with acute MI had an intracoronary thrombus within 4 hours of the onset of symptoms, but only 60% had thrombi 12 to 24 hours later. This observation suggested that the thrombus was quickly dissolved by natural mechanisms after the occlusive event. Further support for the thrombus theory comes from the effectiveness of reperfusion therapies that successfully restore flow through obstructed coronary vessels and significantly reduce mortality.15
The cellular consequences of an acute interruption in blood flow to the myocardium do not occur instantaneously or uniformly. Acute occlusion causes a range of cellular events, depending on the availability and adequacy of collateral blood flow, the relative workload, and the length of time that flow is interrupted. A typical myocardial infarct has several zones composed of cells in various stages of ischemia and death.
Experiments in animal models indicate that complete occlusion of a coronary vessel results in a predictable pattern of cellular dysfunction and death.9 Depletion of ATP in acutely ischemic cells begins immediately, followed within 1 to 2 minutes by an impaired ability to contract. Within 10 minutes, cellular concentrations of ATP fall to half of normal, and irreversible cell injury occurs after 30 to 40 minutes of complete occlusion (Table 18-4). Ischemic necrosis begins in the subendocardial zone and spreads across the ventricular wall toward epicardial surfaces. Epicardial areas are spared for longer periods because they have the greatest collateral network of arterial vessels. The ultimate size of the infarcted tissue depends on the extent, duration, and severity of ischemia. Areas of necrosis may be intermixed with or surrounded by zones of reversibly injured cells that are marginally perfused by collaterals. Injured cells die both from necrosis and from apoptosis.3 Restoring perfusion to potentially salvageable cells is an important focus of treatment. (See Chapter 4 for a discussion of necrosis and apoptosis.)
TABLE 18-4
EVOLUTION OF MORPHOLOGIC CHANGES IN MYOCARDIAL INFARCTION
| TIME | GROSS FEATURES | LIGHT MICROSCOPIC | ELECTRON MICROSCOPIC |
| Reversible Injury | |||
| 0-½ hr | None | None | Relaxation of myofibrils; glycogen loss; mitochondrial swelling |
| Irreversible Injury | |||
| ½-4 hr | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Dark mottling (occasional) | Beginning coagulation necrosis; edema; hemorrhage | |
| 12-24 hr | Dark mottling | Ongoing coagulation necrosis; pyknosis of nuclei; myocyte hypereosinophilia; marginal contraction band necrosis; beginning neutrophilic infiltrate | |
| 1-3 days | Mottling with yellow-tan infarct center | Coagulation necrosis, with loss of nuclei and striations; interstitial infiltrate of neutrophils | |
| 3-7 days | Hyperemic border; central yellow-tan softening | Beginning disintegration of dead myofibers, with dying neutrophils; early phagocytosis of dead cells by macrophages at infarct border | |
| 7-10 days | Maximally yellow-tan and soft, with depressed red-tan margins | Well-developed phagocytosis of dead cells; early formation of fibrovascular granulation tissue at margins | |
| 10-14 days | Red-gray depressed infarct borders | Well-established granulation tissue with new blood vessels and collagen deposition | |
| 2-8 weeks | Gray-white scar, progressive from border toward core of infarct | Increased collagen deposition, with decreased cellularity | |
| >2 mo | Scarring complete | Dense collagenous scar | |
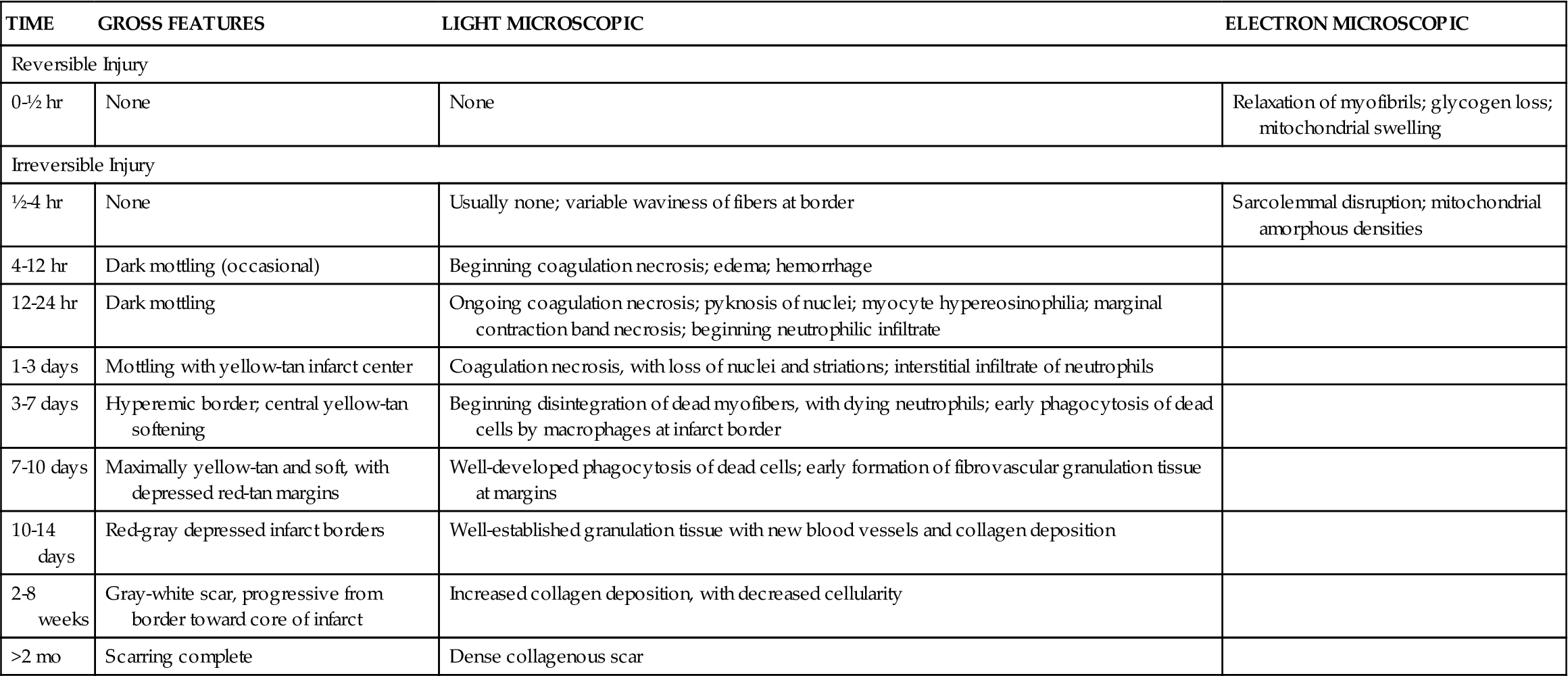
From Kumar V et al: Robbins and Cotran pathologic basis of disease, ed 9, Philadelphia, 2010, Saunders, p 550.
Nearly all infarcts are located in the left ventricular walls. Isolated right ventricular infarction occurs in only 1% to 3% of MIs. Occlusion of the left anterior descending artery causes 40% to 50% of acute MIs, the right coronary artery contributes another 30% to 40%, and the left circumflex contributes 15% to 20%.9 The locations of the resulting infarcts are shown in Table 18-5. It is common for individuals with coronary heart disease to suffer from more than one MI during their lifetime.
TABLE 18-5
LOCATION OF MYOCARDIAL INFARCTION ACCORDING TO CORONARY ARTERY AFFECTED
| ARTERIAL OBSTRUCTION | LOCATION OF INFARCT |
| Left anterior descending (40-50% of infarcts) | Anterior wall of LV near apex Anterior two thirds of interventricular septum |
| Right coronary (30-40% of infarcts) | Posterior wall of LV Posterior one third of interventricular septum |
| Left circumflex (15-20% of infarcts) | Lateral wall of LV |
Data from Kumar V et al: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2010, Saunders, p 551.
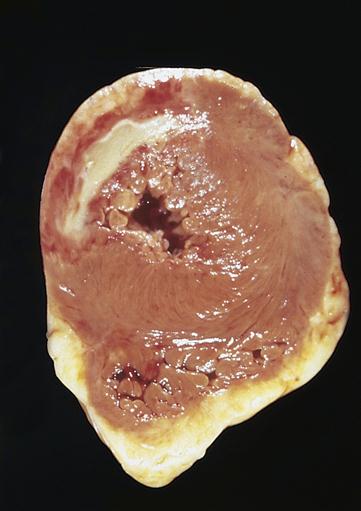
The area of necrosis resulting from MI undergoes a series of morphologic changes as the infarct ages.9 These morphologic changes generally cannot be detected on gross examination until 6 to 12 hours after infarct. After 18 to 24 hours, the area of infarction becomes paler than surrounding tissues. Thereafter, the area of infarction becomes obvious because it turns yellowish and soft with a rim of red vascular connective tissue (Figure 18-7). At 1 to 2 weeks, the necrotic tissue is progressively degraded and cleared from the site. Infarcted myocardium is particularly weakened and susceptible to rupture at this time. By 6 weeks, the necrotic tissue has been replaced by tough fibrous scar tissue.
Diagnosis of MI
The diagnosis of MI is based on three primary indicators: signs and symptoms, electrocardiographic changes, and elevations in the levels of specific marker proteins in the blood. Other diagnostic examinations such as cardiac catheterization, echocardiography, and radionuclide scintigraphy may also be performed to provide additional information (see Chapter 17).
Severe crushing, excruciating chest pain that may radiate to the arm, shoulder, jaw, or back is the harbinger of MI. Pain is commonly accompanied by nausea, vomiting, diaphoresis (sweating), and shortness of breath. In contrast to anginal pain, infarction pain generally lasts more than 15 minutes and is not relieved by rest or nitroglycerin. In some instances, however, the MI is entirely asymptomatic and may elude detection. Asymptomatic MI has been called silent MI and may be detected only serendipitously at a later date. Pain may be difficult to assess in individuals with atypical presentations or a tendency to ignore or deny their symptoms. Thus, although pain is an important indicator of acute ischemia, other clinical information is often needed to correctly distinguish between angina, infarction, and noncardiac sources of pain. Women, the elderly, and patients with diabetic neuropathies more commonly complain of atypical symptoms, including fatigue, nausea, back pain, and abdominal discomfort. Atypical complaints in patients with CHD risk factors should prompt a high suspicion of MI.
Electrocardiographic changes
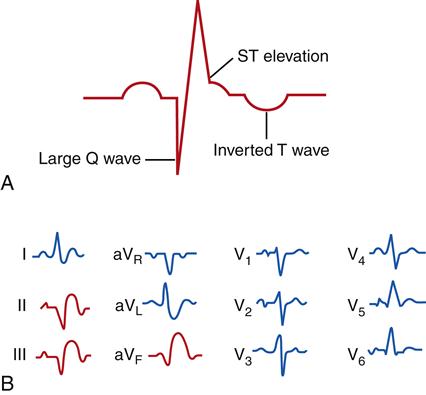
Myocardial ischemia and infarction often result in characteristic changes on ECG waveforms. Injury and ischemia are indicated on the ECG by ST-segment changes. ST-segment elevation is thought to represent acute cellular injury and ischemia. The presence of ST-segment elevation on the ECG indicates that the ischemic injury is ongoing and that efforts to improve perfusion or reduce oxygen demand may be effective in preserving myocardial muscle mass. Until measurements of specific serum markers of infarction are obtained, these patients are regarded as having unstable angina/STEMI in treatment protocols for ACS. Infarcted muscle that is necrotic and no longer electrically active is indicated by the appearance of abnormally deep (>0.1 mV) or wide (>0.03 seconds) Q waves and inverted T waves (Figure 18-8). These changes are very specific for MI and, when present, are diagnostic. Q waves are usually persistent findings, whereas ST-segment and T wave changes may resolve over time. Q waves take time to develop and may not be present in the acute phase of an MI.
Dysrhythmias and the characteristic ST-segment changes that accompany MI are attributed to injured and ischemic cells that have not yet become necrotic. Reversibly injured cells have limited ATP supplies to power membrane pumps and are predisposed to leakage of ions across their cell membranes. Abnormal ion flux may result in continuous current flow even when the heart is at rest. This current leak may be seen on the ECG as ST-segment elevation.
The 12-lead ECG is used to localize the injured region of the left ventricle. Various leads of the 12-lead ECG “view” different regions of the heart. Abnormalities such as Q waves and ST-segment elevation in a particular lead or leads indicate that the damage is localized to the part of the left ventricle “seen” by that lead. MIs may thus be described as anterior, lateral, posterior, septal, inferior, or a combination of these sites. (The 12-lead ECG is described in Chapter 17.)
In some cases, patients presenting with ACS do not have ST elevation on the ECG. They may have ST depression or T wave changes. Some of these patients will develop elevated serum markers indicating MI. These patients have NSTEMI. The infarct size is generally smaller, and the outcomes are better than those in patients with STEMI.13
Serum markers
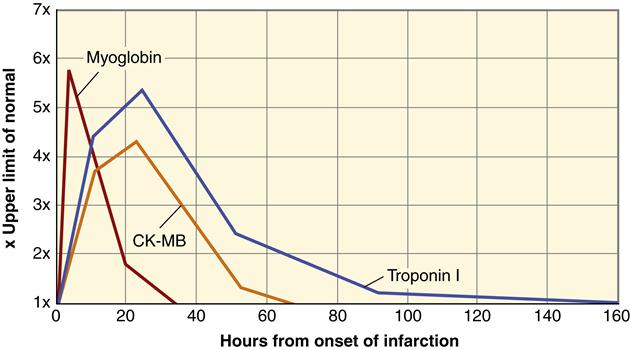
The appearance of certain proteins in the blood after myocardial cell death is a very sensitive and reliable indicator of MI. Myocardial cell death leads to elevated serum levels of myoglobin, troponin, lactate dehydrogenase, and creatine kinase.9 Cardiac cells contain particular forms of these proteins called isoforms. An increase in the concentration of these proteins suggests leakage from fatally damaged cells that have lost plasma membrane integrity. Cardiac biomarkers have a slightly different amino acid sequence than other cell types. In particular, myocytes contain the isoforms CK-MB, troponin I, and troponin T. An elevated level of serum CK-MB is a highly specific indicator of MI and considered to be diagnostic. However, the level of CK-MB remains elevated for only 48 to 72 hours after MI. Two proteins that comprise part of the cardiac cell contractile apparatus, troponins I and T, have become the markers of choice for detecting MI. Cardiac troponin levels become elevated in serum at about the same time as the CK-MB level, but they remain elevated for a longer period. Cardiac troponins I and T are highly sensitive and specific for cardiac cell death but less helpful in detecting new infarction (reinfarction) because levels remain elevated for a prolonged period (Figure 18-9). Cardiac myoglobin levels are elevated in serum very quickly after MI and may be helpful in early detection; however, cardiac myoglobin is less specific than the other markers. All serum markers are useful diagnostically only during the acute period of MI. Patients with ACS who do not develop elevations of these serum markers are diagnosed with unstable angina.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


