Agents Acting at the Neuromuscular Junction and Autonomic Ganglia
The nicotinic acetylcholine (ACh) receptor mediates neurotransmission postsynaptically at the neuromuscular junction and peripheral autonomic ganglia; in the CNS, it largely controls release of neurotransmitters from presynaptic sites. The receptor is called the nicotinic acetylcholine receptor because both the alkaloid nicotine and the neurotransmitter ACh can stimulate the receptor. Distinct subtypes of nicotinic receptors exist at the neuromuscular junction and the ganglia.
THE NICOTINIC ACETYLCHOLINE RECEPTOR
The binding of ACh to the nicotinic ACh receptor initiates an endplate potential (EPP) in muscle or an excitatory postsynaptic potential (EPSP) in peripheral ganglia (see Chapter 8). The nicotinic receptor is the prototype for other pentameric ligand-gated ion channels, which include the receptors for the inhibitory amino acids (γ-aminobutyric acid [GABA] and glycine; see Chapter 14) and serotonin (the 5HT3 receptor) (Figure 11–1).
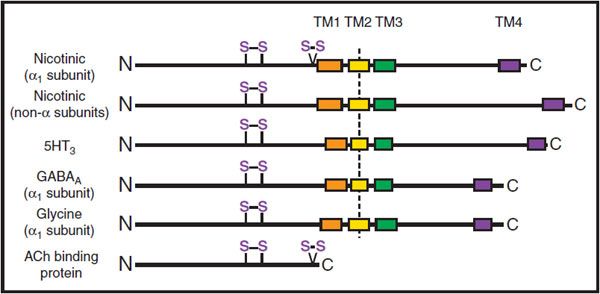
Figure 11–1 Subunit organization of pentameric ligand-gated ion channels and the ACh binding protein. For each receptor, the amino terminal region of ~210 amino acids is found at the extracellular surface. It is then followed by 4 hydrophobic regions that span the membrane (TM1-TM4), leaving the small carboxyl terminus on the extracellular surface. The TM2 region is α-helical, and TM2 regions from each subunit of the pentameric receptor line the internal pore of the receptor. Two disulfide loops at positions 128-142 and 192-193 are found in the α-subunit of the nicotinic receptor. The 128-142 motif is conserved in the family of pentameric receptors; the vicinal cysteines at 192-193 occur only in α-subunits of the nicotinic receptor and in the ACh binding protein.
NICOTINIC RECEPTOR STRUCTURE. Nicotinic receptors of vertebrate skeletal muscle (Nm) are pentamers composed of 4 distinct subunits (α, β, γ, and δ) in the stoichiometric ratio of 2:1:1:1. In mature, innervated muscle endplates, the γ subunit is replaced by ε, a closely related subunit. The individual subunits are ~40% identical in their amino acid sequences. The 5 subunits of the nicotinic acetylcholine receptor are arranged around a pseudo-axis of symmetry to circumscribe a channel. Agonist-binding sites occur at the subunit interfaces; in muscle, only 2 of the 5 subunit interfaces, αγ and αδ, bind ligands (Figure 11–2). Both of the subunits forming the subunit interface contribute to ligand specificity.
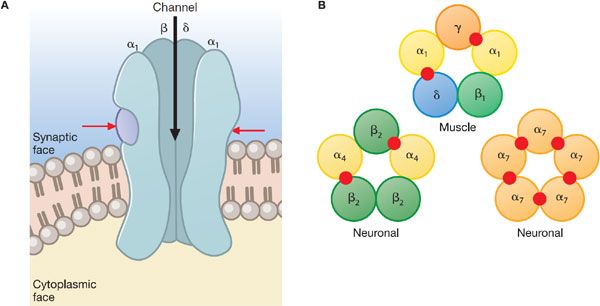
Figure 11–2 Subunit arrangement and molecular structure of the nicotinic acetylcholine receptor. A. Longitudinal view of receptor schematic with the m subunit removed. The remaining subunits, 2 copies of α, 1 of β, and 1 of δ, are shown to surround an internal channel with an outer vestibule and its constriction located deep in the membrane bilayer region. Spans of α-helices with slightly bowed structures form the perimeter of the channel and come from the TM2 region of the linear sequence (Figure 11–1). ACh binding sites, indicated by red arrows, occur at the αγ and αδ (not visible) interfaces. B. Nicotinic receptor subunit arrangement, with examples of subunit assembly. Agonist binding sites (red circles) occur at α subunit-containing interfaces. A total of 17 functional receptor isoforms have been observed in vivo, with different ligand specificity, relative Ca2+/Na+ permeability, and physiological function as determined by their subunit composition. The only isoform found at the neuromuscular junction is that shown here. There are 16 neuronal receptor isoforms found at autonomic ganglia and in the CNS, homo- and hetero-pentamers of α (α2-α10) and β (β2-β4) subunits.
Neuronal nicotinic (Nn) receptors found in ganglia and the CNS also exist as pentamers of 1 or more types of subunits. Subunit types α2 through α10 and β2 through β4 are found in neuronal tissues (see Figure 11–2). Although not all permutations of α and β subunits lead to functional receptors, the diversity in subunit composition is large and exceeds the capacity of ligands to distinguish subtypes on the basis of their selectivity.
NEUROMUSCULAR BLOCKING AGENTS
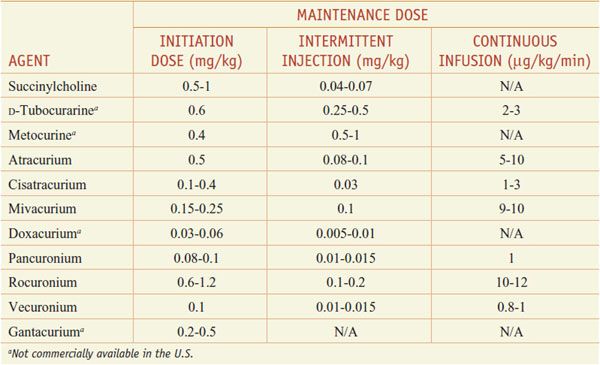
Modern-day neuromuscular blocking agents fall generally into 2 classes, depolarizing and competitive/nondepolarizing. At present, only a single depolarizing agent, succinylcholine (ANECTINE, QUELICIN), is in general clinical use; multiple competitive or nondepolarizing agents are available (see Table 11–2).
Table 11–2
Classification of Neuromuscular Blocking Agents
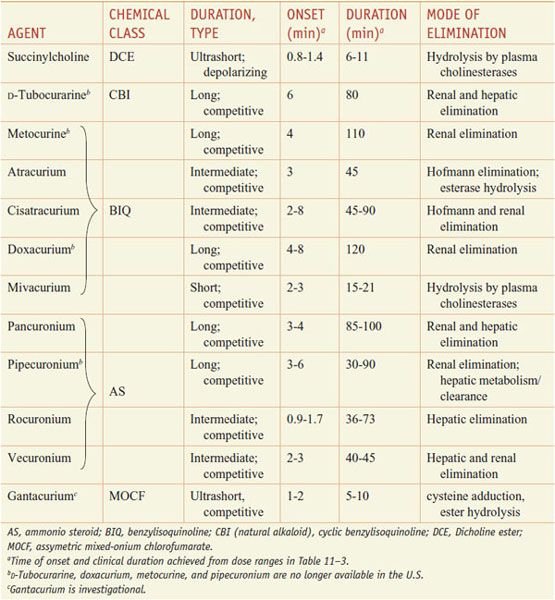
See the 12th edition of the parent text for the history, sources, and chemistry of curare, the prototypical South American arrow poison and neuromuscular blocking agent, the action of which was described by Claude Bernard in the 1850s. The 12th edition also shows the structures of the various classes of depolarizing and competitive neuromuscular blockers in Figure 11–3.
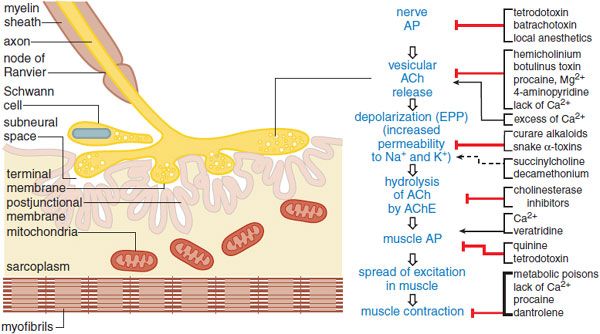
Figure 11–3 A pharmacologist’s view of the motor end plate. The structures of the motor end plate (left side of figure) facilitate the series of physiological events leading from nerve action potential (AP) to skeletal muscle contraction (center column). Pharmacological agents can modify neurotransmission and excitation-contraction coupling at myriad sites (righthand column).  enhancement;
enhancement;  blockade;
blockade;  depolarization and phase II block.
depolarization and phase II block.
Pharmacological Effects
SKELETAL MUSCLE. Competitive antagonists bind the nicotinic ACh receptor of the motor endplate and thereby competitively block the binding of ACh. The depolarizing agents, such as succinylcholine, depolarize the membrane by opening channels in the same manner as ACh. However, they persist longer at the neuromuscular junction primarily because of their resistance to AChE. The depolarization is thus longer lasting, resulting in a brief period of repetitive excitation that may elicit transient and repetitive muscle excitation (fasciculations), followed by block of neuromuscular transmission and flaccid paralysis (called phase I block).
The block arises because, after an initial opening, peri-junctional Na+ channels close and will not reopen until the endplate is repolarized. At this point, neural release of ACh results in the binding of ACh to receptors on an already depolarized endplate. These closed perijunctional channels keep the depolarization signal from affecting downstream channels and effectively shield the rest of the muscle from activity at the motor endplate. The characteristics of depolarization and competitive blockade are contrasted in Table 11–3.
Table 11–3
Dosing Ranges for Neuromuscular Blocking Agents
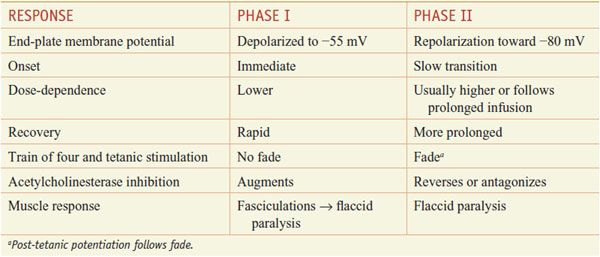
Under clinical conditions, with increasing concentrations of succinylcholine and over time, the block may convert slowly from a depolarizing phase I block to a nondepolarizing, phase II block. While the response to peripheral stimulation during phase II block resembles that of the competitive agents, reversal of phase II block by administration of anticholinesterase (anti-ChE) agents (e.g., with neostigmine) is difficult to predict and should be undertaken with much caution. The characteristics of phase I and phase II blocks are shown in Table 11–1.
Table 11–1
Clinical Responses and Monitoring of Phase I and Phase II Neuromuscular Blockade by Succinylcholine Infusion
Sequence and Characteristics of Paralysis. Following intravenous injection of an appropriate dose of a competitive blocking agent, motor weakness progresses to a total flaccid paralysis. Small, rapidly moving muscles such as those of the eyes, jaw, and larynx relax before those of the limbs and trunk. Ultimately, the intercostal muscles and finally the diaphragm are paralyzed, and respiration then ceases. Recovery of muscles usually occurs in the reverse order to that of their paralysis, and thus the diaphragm ordinarily is the first muscle to regain function.
After a single intravenous dose of 10-30 mg of succinylcholine, muscle fasciculations, particularly over the chest and abdomen, occur briefly; then relaxation occurs within 1 min, becomes maximal within 2 min, and generally disappears within 5 min. Transient apnea usually occurs at the time of maximal effect. Muscle relaxation of longer duration is achieved by continuous intravenous infusion. After infusion is discontinued, the effects of the drug usually disappear rapidly because of its efficient hydrolysis by plasma and hepatic butyrylcholinesterase. Muscle soreness may follow the administration of succinylcholine.
During prolonged depolarization, muscle cells may lose significant quantities of K+ and gain Na+, Cl–, and Ca2+. In patients with extensive injury to soft tissues, the efflux of K+ following continued administration of succinylcholine can be life threatening. There are many conditions for which succinylcholine administration is contraindicated or should be undertaken with great caution. The change in the nature of the blockade produced by succinylcholine (from phase I to phase II) presents an additional complication with long-term infusions.
CNS. Tubocurarine and other quaternary neuromuscular blocking agents are virtually devoid of central effects following ordinary clinical doses because of their inability to penetrate the blood-brain barrier.
AUTONOMIC GANGLIA AND MUSCARINIC SITES. Neuromuscular blocking agents show variable potencies in producing ganglionic blockade. Ganglionic blockade by tubocurarine and other stabilizing drugs is reversed or antagonized by anti-ChE agents.
Clinical doses of tubocurarine produce partial blockade both at autonomic ganglia and at the adrenal medulla, which results in a fall in blood pressure and tachycardia. Pancuronium shows less ganglionic blockade at common clinical doses. Atracurium, vecuronium, doxacurium, pipecuronium, mivacurium, and rocuronium are even more selective. The maintenance of cardiovascular reflex responses usually is desired during anesthesia. Pancuronium has a vagolytic action, presumably from blockade of muscarinic receptors, which leads to tachycardia.
Of the depolarizing agents, succinylcholine at doses producing neuromuscular relaxation rarely causes effects attributable to ganglionic blockade. However, cardiovascular effects are sometimes observed, probably owing to the successive stimulation of vagal ganglia (manifested by bradycardia) and sympathetic ganglia (resulting in hypertension and tachycardia).
MAST CELLS AND HISTAMINE RELEASE. Tubocurarine produces typical histamine-like wheals when injected intracutaneously or intraarterially in humans, and some clinical responses to neuromuscular blocking agents (e.g., bronchospasm, hypotension, excessive bronchial and salivary secretion) appear to be caused by the release of histamine. Succinylcholine, mivacurium, and atracurium also cause histamine release, but to a lesser extent unless administered rapidly. The ammonio steroids, pancuronium, vecuronium, pipecuronium, and rocuronium, have even less tendency to release histamine after intradermal or systemic injection. Histamine release typically is a direct action of the muscle relaxant on the mast cell rather than IgE-mediated anaphylaxis.
ABSORPTION, DISTRIBUTION, AND ELIMINATION
Quaternary ammonium neuromuscular blocking agents are very poorly absorbed from the GI tract. Absorption is quite adequate from intramuscular sites. Rapid onset is achieved with intravenous administration. The more potent agents must be given in lower concentrations, and diffusional requirements slow their rate of onset.
When long-acting competitive blocking agents such as D-tubocurarine and pancuronium are administered, blockade may diminish after 30 min owing to redistribution of the drug, yet residual blockade and plasma levels of the drug persist. Subsequent doses show diminished redistribution. Long-acting agents may accumulate with multiple doses.
The ammonio steroids contain ester groups that are hydrolyzed in the liver. Typically, the metabolites have about one-half the activity of the parent compound and contribute to the total relaxation profile. Ammonio steroids of intermediate duration of action, such as vecuronium and rocuronium (Table 11–2), are cleared more rapidly by the liver than is pancuronium. The more rapid decay of neuromuscular blockade with compounds of intermediate duration argues for sequential dosing of these agents rather than administering a single dose of a long-duration neuromuscular blocking agent.
Atracurium is converted to less active metabolites by plasma esterases and spontaneous Hofmann elimination. Cisatracurium is also subject to this spontaneous degradation. Because of these alternative routes of metabolism, atracurium and cisatracurium do not exhibit an increased t1/2 in patients with impaired renal function and therefore are good choices in this setting.
The extremely brief duration of action of succinylcholine is due largely to its rapid hydrolysis by the butyrylcholinesterase synthesized by the liver and found in the plasma. Among the occasional patients who exhibit prolonged apnea following the administration of succinylcholine or mivacurium, most have an atypical plasma cholinesterase or a deficiency of the enzyme owing to allelic variations, hepatic or renal disease, or a nutritional disturbance; however, in some, the enzymatic activity in plasma is normal.
Gantacurium is degraded by 2 chemical mechanisms, a rapid cysteine adduction and a slower hydrolysis of the ester bond adjacent to the chlorine. Both processes are purely chemical and hence not dependent on enzymatic activities. The adduction process has a t1/2 of 1-2 min and is likely the basis for the ultrashort duration of action of gantacurium. Administration of exogenous cysteine, which may have excitotoxic side effects, can accelerate the antagonism of gantacurium-induced neuromuscular blockade.
CLINICAL PHARMACOLOGY
CHOICE OF AGENT
Therapeutic selection of a neuromuscular blocking agent should be based on achieving a pharmacokinetic profile consistent with the duration of the interventional procedure and minimizing cardiovascular compromise or other side effects, with attention to drug-specific modes of elimination in patients with renal or hepatic failure (see Table 11–2).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


