Adrenergic Agonists and Antagonists
Catecholamines and Sympathomimetic Drugs
Catecholamines and sympathomimetic drugs are classified as direct-acting, indirect-acting, or mixed-acting sympathomimetics (Figure 12–1).
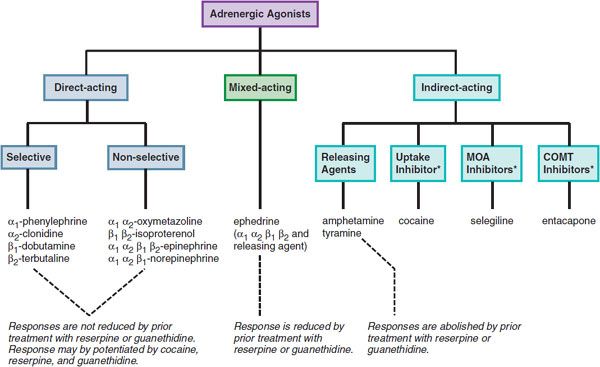
Figure 12–1 Classification of adrenergic receptor agonists (sympathomimetic amines) and drugs that produce sympathomimetic-like effects. For each category, a prototypical drug is shown. (*Not actually sympathetic drugs but produce sympathomimetic-like effects.)
Direct-acting sympathomimetic drugs act directly on 1 or more of the adrenergic receptors. These agents may exhibit considerable selectivity for a specific receptor subtype (e.g., phenylephrine for α1, terbutaline for β2) or may have no or minimal selectivity and act on several receptor types (e.g., epinephrine for α1, α2, β1, β2, and β3 receptors; norepinephrine for α1, α2, and β1 receptors).
Indirect-acting drugs increase the availability of norepinephrine (NE) or epinephrine (EPI) to stimulate adrenergic receptors by several mechanisms:
• By releasing or displacing NE from sympathetic nerve varicosities
• By inhibiting the transport of NE into sympathetic neurons (e.g., cocaine), thereby increasing the dwell time of the transmitter at the receptor
• By blocking the metabolizing enzymes, monoamine oxidase (MAO) (e.g., pargyline) or catechol-O-methyltransferase (COMT) (e.g., entacapone), effectively increasing transmitter supply
Drugs that indirectly release NE and also directly activate receptors are referred to as mixed-acting sympathomimetic drugs [e.g., ephedrine, dopamine (DA)].
A feature of direct-acting sympathomimetic drugs is that their responses are not reduced by prior treatment with reserpine or guanethidine, which deplete NE from sympathetic neurons. After transmitter depletion, the actions of direct-acting sympathomimetic drugs actually may increase because the loss of the neurotransmitter induces compensatory changes that upregulate receptors or enhance the signaling pathway. In contrast, the responses of indirect-acting sympathomimetic drugs (e.g., amphetamine, tyramine) are abolished by prior treatment with reserpine or guanethidine. The cardinal feature of mixed-acting sympathomimetic drugs is that their effects are blunted, but not abolished, by prior treatment with reserpine or guanethidine.
Because the actions of NE are more pronounced on α and β1 receptors than on β2 receptors, many non-catecholamines that release NE have predominantly α receptor–mediated and cardiac effects. However, certain non-catecholamines with both direct and indirect effects on adrenergic receptors show significant β2 activity and are used clinically for these effects. Thus, ephedrine, although dependent on release of NE for some of its effects, relieves bronchospasm by its action on β2 receptors in bronchial smooth muscle, an effect not seen with NE. Moreover, some non-catecholamines (e.g., phenylephrine) act primarily and directly on target cells. It therefore is impossible to predict precisely the effects of non-catecholamines solely on their ability to provoke NE release.
CHEMISTRY AND STRUCTURE-ACTIVITY RELATIONSHIP OF SYMPATHOMIMETIC AMINES. β-Phenylethylamine (Table 12–1) can be viewed as the parent compound of the sympathomimetic amines, consisting of a benzene ring and an ethylamine side chain. The structure permits substitutions to be made on the aromatic ring, the α- and β-carbon atoms, and the terminal amino group to yield a variety of compounds with sympathomimetic activity. NE, EPI, DA, isoproterenol, and a few other agents have hydroxyl groups substituted at positions 3 and 4 of the benzene ring. Since o-dihydroxybenzene is also known as catechol, sympathomimetic amines with these hydroxyl substitutions in the aromatic ring are termed catecholamines.
Table 12–1
Chemical Structures and Major Effects of Some Sympathomimetic Drugs
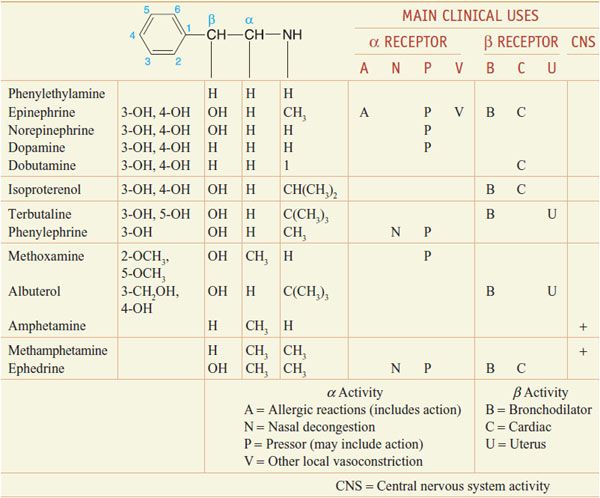
Many directly acting sympathomimetic drugs influence both α and β receptors, but the ratio of activities varies among drugs in a continuous spectrum from predominantly α activity (phenylephrine) to predominantly β activity (isoproterenol) (see Table 12–1).
Catecholamines have only a brief duration of action and are ineffective when administered orally, because they are rapidly inactivated in the intestinal mucosa and in the liver before reaching the systemic circulation (see Chapter 8). Compounds without 1 or both hydroxyl substituents are not acted on by COMT, and their oral effectiveness and duration of action are enhanced.
PHYSIOLOGICAL BASIS OF ADRENERGIC RECEPTOR FUNCTION. An important factor in the response of any cell or organ to sympathomimetic amines is the density and proportion of α and β adrenergic receptors. For example, NE has relatively little capacity to increase bronchial airflow, since the receptors in bronchial smooth muscle are largely of the β2 subtype. In contrast, isoproterenol and EPI are potent bronchodilators. Cutaneous blood vessels physiologically express almost exclusively α receptors; thus, NE and EPI cause constriction of such vessels, whereas isoproterenol has little effect. The smooth muscle of blood vessels that supply skeletal muscles has both β2 and α receptors; activation of β2 receptors causes vasodilation, and stimulation of α receptors constricts these vessels. In such vessels, the threshold concentration for activation of β2 receptors by EPI is lower than that for α receptors, but when both types of receptors are activated at high concentrations of EPI, the response to α receptors predominates. Physiological concentrations of EPI primarily cause vasodilation.
The ultimate response of a target organ to sympathomimetic amines is dictated by the direct effects of the agents and the reflex homeostatic adjustments of the organism. Many sympathomimetic amines produce a rise in arterial blood pressure caused by stimulation of vascular α adrenergic receptors. This stimulation elicits compensatory reflexes that are mediated by the carotid–aortic baroreceptor system. As a result, sympathetic tone is diminished and vagal tone is enhanced; each of these responses leads to slowing of the heart rate. Conversely, when a drug (e.g., a β2 agonist) lowers mean blood pressure at the mechanoreceptors of the carotid sinus and aortic arch, the baroreceptor reflex works to restore pressure by reducing parasympathetic (vagal) outflow from the CNS to the heart, and increasing sympathetic outflow to the heart and vessels.
FALSE-TRANSMITTER CONCEPT. This hypothesis is a possible explanation for some of the effects of MAO inhibitors. Phenylethylamines normally are synthesized in the GI tract as a result of the action of bacterial tyrosine decarboxylase. The tyramine formed in this fashion usually is oxidatively deaminated in the GI tract and the liver. However, when an MAO inhibitor is administered, tyramine may be absorbed systemically and transported into sympathetic nerve terminals, where its catabolism again is prevented because of the inhibition of MAO at this site; the tyramine then is β-hydroxylated to octopamine, which is stored in vesicles. As a consequence, NE gradually is displaced by octopamine, and stimulation of the nerve terminal results in the release of a relatively small amount of NE along with a fraction of octopamine. The latter amine has relatively little ability to activate either α or β receptors. Thus, a functional impairment of sympathetic transmission parallels long-term administration of MAO inhibitors.
Despite such functional impairment, patients who have received MAO inhibitors may experience severe hypertensive crises if they ingest cheese, beer, or red wine. These and related foods, which are produced by fermentation, contain a large quantity of tyramine, and to a lesser degree, other phenylethylamines. When GI and hepatic MAO are inhibited, the large quantity of tyramine that is ingested is absorbed rapidly and reaches the systemic circulation in high concentration. A massive and precipitous release of NE can result, with consequent hypertension that can be severe enough to cause myocardial infarction or a stroke (see Chapter 15).
ENDOGENOUS CATECHOLAMINES
EPINEPHRINE
EPI (adrenaline) is a potent stimulant of both α and β adrenergic receptors. Most of the responses listed in Table 8–1 are seen after injection of EPI, although the occurrence of sweating, piloerection, and mydriasis depends on the physiological state of the subject. Particularly prominent are the actions on the heart and on vascular and other smooth muscle.
BLOOD PRESSURE. EPI is one of the most potent vasopressor drugs known. If a pharmacological dose is given rapidly by an intravenous route, it evokes a characteristic effect on blood pressure, which rises rapidly to a peak that is proportional to the dose. The increase in systolic pressure is greater than the increase in diastolic pressure, so that the pulse pressure increases. As the response wanes, the mean pressure may fall below normal before returning to control levels.
The mechanism of the rise in blood pressure due to EPI is:
• A direct myocardial stimulation that increases the strength of ventricular contraction (positive inotropic action)
• An increased heart rate (positive chronotropic action)
• Vasoconstriction in many vascular beds—especially in the precapillary resistance vessels of skin, mucosa, and kidney—along with marked constriction of the veins
The pulse rate, at first accelerated, may be slowed markedly at the height of the rise of blood pressure by compensatory vagal discharge (baroreceptor reflex). Small doses of EPI (0.1 μg/kg) may cause the blood pressure to fall. The depressor effect of small doses and the biphasic response to larger doses are due to greater sensitivity to EPI of vasodilator β2 receptors than of constrictor α receptors.
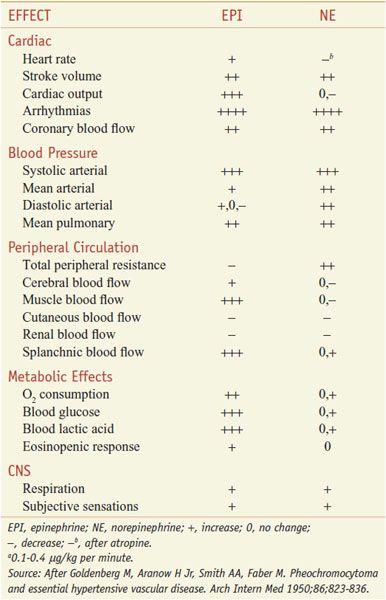
Absorption of EPI after subcutaneous injection is slow due to local vasoconstrictor action; the effects of doses as large as 0.5-1.5 mg can be duplicated by intravenous infusion at a rate of 10-30 μg/min. There is a moderate increase in systolic pressure due to increased cardiac contractile force and a rise in cardiac output (Figure 12–2). Peripheral resistance decreases, owing to a dominant action on β2 receptors of vessels in skeletal muscle, where blood flow is enhanced; as a consequence, diastolic pressure usually falls. Because the mean blood pressure is not, as a rule, greatly elevated, compensatory baroreceptor reflexes do not appreciably antagonize the direct cardiac actions. Heart rate, cardiac output, stroke volume, and left ventricular work per beat are increased as a result of direct cardiac stimulation and increased venous return to the heart, which is reflected by an increase in right atrial pressure. The details of the effects of intravenous infusion of EPI, NE, and isoproterenol in humans are compared in Table 12–2 and Figure 12–2.
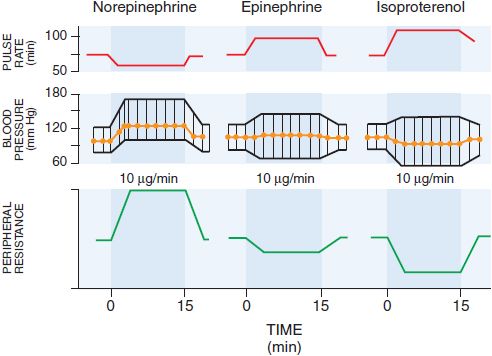
Figure 12–2 Effects of intravenous infusion of norepinephrine, epinephrine, or isoproterenol in humans. (Modified from Allwood MJ, Cobbold AF, Ginsberg J. Peripheral vascular effects of noradrenaline, isopropylnoradrenaline, and dopamine. Br Med Bull, 1963;19:132-136. With permission from Oxford University Press.)
Table 12–2
Comparative Effects of Infusion of Epinephrine and Norepinephrine in Humansa
VASCULAR EFFECTS. In the vasculature, EPI acts chiefly on the smaller arterioles and precapillary sphincters, although veins and large arteries also respond to the drug. Various vascular beds react differently, which results in a substantial redistribution of blood flow. Injected EPI markedly decreases cutaneous blood flow, constricting precapillary vessels and small venules. Cutaneous vasoconstriction accounts for a marked decrease in blood flow in the hands and feet. Blood flow to skeletal muscles is increased by therapeutic doses in humans. This is due in part to a powerful β2-mediated vasodilator action that is only partially counterbalanced by a vasoconstrictor action on the α receptors that also are present in the vascular bed.
The effect of EPI on cerebral circulation is related to systemic blood pressure. In usual therapeutic doses, the drug has relatively little constrictor action on cerebral arterioles. Indeed, autoregulatory mechanisms tend to limit the increase in cerebral blood flow caused by increased blood pressure.
Doses of EPI that have little effect on mean arterial pressure consistently increase renal vascular resistance and reduce renal blood flow by as much as 40%. Since the glomerular filtration rate is only slightly and variably altered, the filtration fraction is consistently increased. Excretion of Na+, K+, and Cl– is decreased; urine volume may be increased, decreased, or unchanged. Maximal tubular reabsorptive and excretory capacities are unchanged. The secretion of renin is increased as a consequence of a direct action of EPI on β1 receptors in the juxtaglomerular apparatus. Arterial and venous pulmonary pressures are raised. Although direct pulmonary vasoconstriction occurs, redistribution of blood from the systemic to the pulmonary circulation, due to constriction of the more powerful musculature in the systemic great veins, doubtless plays an important part in the increase in pulmonary pressure. Very high concentrations of EPI may cause pulmonary edema precipitated by elevated pulmonary capillary filtration pressure and possibly by “leaky” capillaries.
Coronary blood flow is enhanced by EPI or by cardiac sympathetic stimulation under physiological conditions. The increased flow, which occurs even with doses that do not increase the aortic blood pressure, is the result of 2 factors. The first is the increased relative duration of diastole at higher heart rates; this is partially offset by decreased blood flow during systole because of more forceful contraction of the surrounding myocardium and an increase in mechanical compression of the coronary vessels. The increased flow during diastole is further enhanced if aortic blood pressure is elevated by EPI; as a consequence, total coronary flow may be increased. The second factor is a metabolic dilator effect that results from the increased strength of contraction and myocardial O2 consumption due to the direct effects of EPI on cardiac myocytes. This vasodilation is mediated in part by adenosine released from cardiac myocytes, which tends to override a direct vasoconstrictor effect of EPI that results from activation of α receptors in coronary vessels.
CARDIAC EFFECTS. EPI is a powerful cardiac stimulant. It acts directly on the predominant β1 receptors of the myocardium and of the cells of the pacemaker and conducting tissues; β2, β3, and α receptors also are present in the heart. Direct responses to EPI include increases in contractile force, accelerated rate of rise of isometric tension, enhanced rate of relaxation, decreased time to peak tension, increased excitability, acceleration of the rate of spontaneous beating, and induction of automaticity in specialized regions of the heart. In accelerating the heart, EPI preferentially shortens systole so that the duration of diastole usually is not reduced.
EPI normally shortens the refractory period of the human atrioventricular (AV) node by direct effects on the heart, although doses of EPI that slow the heart through reflex vagal discharge may indirectly tend to prolong it. EPI also decreases the grade of AV block that occurs as a result of disease, drugs, or vagal stimulation. Depression of sinus rate and AV conduction by vagal discharge probably plays a part in EPI-induced ventricular arrhythmias, because various drugs that block the vagal effect confer some protection. The actions of EPI in enhancing cardiac automaticity are effectively antagonized by β receptor antagonists such as propranolol. However, α1 receptors exist in most regions of the heart, and their activation prolongs the refractory period and strengthens myocardial contractions. Cardiac arrhythmias have been seen in patients after inadvertent intravenous administration of conventional subcutaneous doses of EPI. EPI as well as other catecholamines may cause myocardial cell death, particularly after intravenous infusions. Acute toxicity is associated with contraction band necrosis and other pathological changes; prolonged sympathetic stimulation of the heart, such as in congestive cardiomyopathy, may promote apoptosis of cardiomyocytes.
EFFECTS ON SMOOTH MUSCLES. The effects of EPI on the smooth muscles of different organs and systems depend on the type of adrenergic receptor in the muscle (see Table 8–1). In general, EPI relaxes GI smooth muscle due to activation of both α and β receptors. Intestinal tone and the frequency and amplitude of spontaneous contractions are reduced. The stomach usually is relaxed and the pyloric and ileocecal sphincters are contracted, but these effects depend on the preexisting tone of the muscle. If tone already is high, EPI causes relaxation; if low, contraction.
The responses of uterine muscle to EPI vary with species, phase of the sexual cycle, state of gestation, and dose given. During the last month of pregnancy and at parturition, EPI inhibits uterine tone and contractions (see Chapter 66). EPI relaxes the detrusor muscle of the bladder as a result of activation of β receptors and contracts the trigone and sphincter muscles owing to its α-agonist activity. This can result in hesitancy in urination and may contribute to retention of urine in the bladder. Activation of smooth muscle contraction in the prostate promotes urinary retention.
RESPIRATORY EFFECTS. EPI has a powerful bronchodilator action, most evident when bronchial muscle is contracted because of disease, as in bronchial asthma, or in response to drugs or various autacoids.
The beneficial effects of EPI in asthma also may arise from inhibition of antigen-induced release of inflammatory mediators from mast cells, and to a lesser extent from diminution of bronchial secretions and congestion within the mucosa. Inhibition of mast cell secretion is mediated by β2 receptors, while the effects on the mucosa are mediated by α receptors. however, glucocorticoids have much more profound anti-inflammatory effects (see Chapters 33 and 34).
EFFECTS ON THE CNS. EPI, a polar compound, penetrates poorly into the CNS and hence is not a powerful CNS stimulant. While the drug may cause restlessness, apprehension, headache, and tremor in many persons, these effects in part may be secondary to the effects of EPI on the cardiovascular system, skeletal muscles, and intermediary metabolism; that is, they may be the result of somatic manifestations of anxiety.
METABOLIC EFFECTS. EPI elevates the concentrations of glucose and lactate in blood (see Chapter 8). Insulin secretion is inhibited through an interaction with α2 receptors and is enhanced by activation of α2 receptors; the predominant effect seen with EPI is inhibition. Glucagon secretion is enhanced by an action on the β receptors of the α cells of pancreatic islets. EPI also decreases the uptake of glucose by peripheral tissues. EPI stimulates glycogenolysis in most tissues through β receptors. EPI raises the concentration of free fatty acids in blood by stimulating β receptors in adipocytes. The calorigenic action of EPI (increase in metabolism) is reflected in humans by an increase of 20-30% in O2 consumption after conventional doses.
MISCELLANEOUS EFFECTS. EPI reduces circulating plasma volume by loss of protein-free fluid to the extracellular space, thereby increasing hematocrit and plasma protein concentration. EPI rapidly increases the number of circulating polymorphonuclear leukocytes, likely due to β receptor–mediated demargination of these cells. EPI accelerates blood coagulation and promotes fibrinolysis. EPI stimulates lacrimation and a scanty mucus secretion from salivary glands. EPI also acts directly on white, fast-twitch muscle fibers to prolong the active state, thereby increasing peak tension.
EPI promotes a fall in plasma K+, largely due to stimulation of K+ uptake into cells, particularly skeletal muscle, due to activation of β2 receptors. This is associated with decreased renal K+ excretion.
ABSORPTION, FATE, AND EXCRETION. EPI is ineffective after oral administration. Absorption from subcutaneous tissues occurs relatively slowly. Absorption is more rapid after intramuscular injection. When concentrated solutions are nebulized and inhaled, the actions of the drug largely are restricted to the respiratory tract; however, systemic reactions such as arrhythmias may occur, particularly if larger amounts are used. EPI is rapidly inactivated in the liver by COMT and MAO (see Figure 8–7 and Table 8–4).
EPI is available in a variety of formulations geared for different routes of administration, including self-administration for anaphylactic reactions (EpiPen). EPI is unstable in alkaline solution; when exposed to air or light, it turns pink from oxidation to adrenochrome and then brown from formation of polymers. EPI injection is available in 1 mg/mL (1:1000), 0.1 mg/mL (1:10,000), and 0.5 mg/mL (1:2000) solutions. Subcutaneous dose ranges from 0.3-0.5 mg. The intravenous route is used cautiously if an immediate and reliable effect is mandatory. If the solution is given by vein, it must be adequately diluted and injected very slowly.
TOXICITY, ADVERSE EFFECTS, AND CONTRAINDICATIONS. EPI may cause restlessness, throbbing headache, tremor, and palpitations. The effects rapidly subside with rest, quiet, recumbency, and reassurance. More serious reactions include cerebral hemorrhage and cardiac arrhythmias. The use of large doses or the accidental, rapid intravenous injection of EPI may result in cerebral hemorrhage from the sharp rise in blood pressure. Angina may be induced by EPI in patients with coronary artery disease. The use of EPI generally is contraindicated in patients who are receiving nonselective β receptor antagonists, because its unopposed actions on vascular α1 receptors may lead to severe hypertension and cerebral hemorrhage.
THERAPEUTIC USES. A major use of EPI is to provide rapid, emergency relief of hypersensitivity reactions, including anaphylaxis, to drugs and other allergens. EPI also is used to prolong the action of local anesthetics, presumably by decreasing local blood flow (see Chapter 20). Its cardiac effects may be of use in restoring cardiac rhythm in patients with cardiac arrest due to various causes. It also is used as a topical hemostatic agent on bleeding surfaces such as in the mouth or in bleeding peptic ulcers during endoscopy of the stomach and duodenum. Inhalation of EPI may be useful in the treatment of post-intubation and infectious croup.
NOREPINEPHRINE
Norepinephrine (levarterenol, l-noradrenaline, NE) is a major chemical mediator liberated by mammalian postganglionic sympathetic nerves (see Table 12–1).
NE constitutes 10-20% of the catecholamine content of human adrenal medulla and as much as 97% in some pheochromocytomas.
PHARMACOLOGICAL PROPERTIES. The pharmacological actions of NE and EPI are compared in Table 12–2. Both drugs are direct agonists on effector cells; their actions differ mainly in the ratio of their effectiveness in stimulating α and β2 receptors. They are approximately equipotent in stimulating β1 receptors. NE is a potent α agonist, has relatively little action on β2 receptors, and is somewhat less potent than EPI on the α receptors of most organs.
ABSORPTION, FATE, AND EXCRETION. NE, like EPI, is ineffective when given orally and is absorbed poorly from sites of subcutaneous injection. It is rapidly inactivated in the body by uptake and the actions of COMT and MAO. Small amounts normally are found in the urine. The excretion rate may be greatly increased in patients with pheochromocytoma.
CARDIOVASCULAR EFFECTS. In response to infused NE (see Figure 12–2), systolic and diastolic pressures, and usually pulse pressure, are increased. Cardiac output is unchanged or decreased, and total peripheral resistance is raised. Compensatory vagal reflex activity slows the heart, overcoming a direct cardioaccelerator action, and stroke volume is increased. The peripheral vascular resistance increases in most vascular beds, and renal blood flow is reduced. NE constricts mesenteric vessels and reduces splanchnic and hepatic blood flow. Coronary flow usually is increased, probably owing both to indirectly induced coronary dilation, as with EPI, and to elevated blood pressure. Although generally a poor β2 receptor agonist, NE may increase coronary blood flow directly by stimulating β2receptors on coronary vessels. Patients with Prinzmetal variant angina pectoris may be supersensitive to the α adrenergic vasoconstrictor effects of NE.
ADVERSE EFFECTS AND PRECAUTIONS. The untoward effects of NE are similar to those of EPI, although there typically is greater elevation of blood pressure with NE. Care must be taken that necrosis and sloughing do not occur at the site of intravenous injection owing to extravasation of the drug. Impaired circulation at injection sites may be relieved by infiltrating the area with phentolamine, an α receptor antagonist. Blood pressure must be determined frequently during the infusion and particularly during adjustment of the rate of the infusion. Reduced blood flow to organs such as kidney and intestines is a constant danger with the use of NE.
THERAPEUTIC USES AND STATUS. NE (LEVOPHED, others) is used as a vasoconstrictor to raise or support blood pressure under certain intensive care conditions (discussed below).
DOPAMINE
DA (see Table 12–1) is the immediate metabolic precursor of NE and EPI; it is a central neurotransmitter important in the regulation of movement (see Chapters 14, 16, and 22) and possesses important intrinsic pharmacological properties. In the periphery, it is synthesized in epithelial cells of the proximal tubule and is thought to exert local diuretic and natriuretic effects. DA is a substrate for both MAO and COMT and thus is ineffective when administered orally.
PHARMACOLOGICAL PROPERTIES.
Cardiovascular Effects. The cardiovascular effects of DA are mediated by several distinct types of receptors that vary in their affinity for DA (see Chapter 13). At low concentrations, the primary interaction of DA is with vascular D1 receptors, especially in the renal, mesenteric, and coronary beds. By activating adenylyl cyclase and raising intracellular concentrations of cyclic AMP, D1 receptor stimulation leads to vasodilation. Infusion of low doses of DA causes an increase in glomerular filtration rate, renal blood flow, and Na+ excretion. Activation of D1 receptors on renal tubular cells decreases Na+ transport by cyclic AMP-dependent and cyclic AMP-independent mechanisms. The renal tubular actions of DA that cause natriuresis may be augmented by the increase in renal blood flow and the small increase in the glomerular filtration rate that follow its administration. The resulting increase in hydrostatic pressure in the peritubular capillaries and reduction in oncotic pressure may contribute to diminished reabsorption of Na+ by the proximal tubular cells. As a consequence, DA has pharmacologically appropriate effects in the management of states of low cardiac output associated with compromised renal function, such as severe congestive heart failure.
At higher concentrations, DA exerts a positive inotropic effect on the myocardium, acting on β1 adrenergic receptors. DA also causes the release of NE from nerve terminals, which contributes to its effects on the heart. Tachycardia is less prominent during infusion of DA than of isoproterenol. DA usually increases systolic blood pressure and pulse pressure and either has no effect on diastolic blood pressure or increases it slightly. Total peripheral resistance usually is unchanged when low or intermediate doses of DA are given. At high concentrations, DA activates vascular α1 receptors, leading to more general vasoconstriction.
PRECAUTIONS, ADVERSE REACTIONS, AND CONTRAINDICATIONS. Before DA is administered to patients in shock, hypovolemia should be corrected by transfusion of whole blood, plasma, or other appropriate fluid. Untoward effects due to overdosage generally are attributable to excessive sympathomimetic activity (although this also may be the response to worsening shock). Nausea, vomiting, tachycardia, anginal pain, arrhythmias, headache, hypertension, and peripheral vasoconstriction may be encountered during DA infusion. Extravasation of large amounts of DA during infusion may cause ischemic necrosis and sloughing. Rarely, gangrene of the fingers or toes has followed prolonged infusion of the drug. DA should be avoided if the patient has received an MAO inhibitor. Careful adjustment of dosage also is necessary in patients who are taking tricyclic antidepressants.
THERAPEUTIC USES. DA is used in the treatment of severe congestive heart failure, particularly in patients with oliguria and low or normal peripheral vascular resistance. The drug may improve physiological parameters in the treatment of cardiogenic and septic shock. While DA may acutely improve cardiac and renal function in severely ill patients with chronic heart disease or renal failure, there is relatively little evidence supporting long-term benefit in clinical outcome.
Dopamine hydrochloride is used only intravenously, administered at a rate of 2-5 μg/kg/min; this rate may be increased gradually up to 20-50 μg/kg/min as necessary. During the infusion, patients require clinical assessment of myocardial function, perfusion of vital organs such as the brain, and the production of urine. Reduction in urine flow, tachycardia, or the development of arrhythmias may be indications to slow or terminate the infusion. The duration of action of DA is brief, and hence the rate of administration can be used to control the intensity of effect.
Related drugs include fenoldopam and dopexamine. Fenoldopam (CORLOPAM, others), a benzazepine derivative, is a rapidly acting vasodilator used for control of severe hypertension (e.g., malignant hypertension with end-organ damage) in hospitalized patients for ≤48 h. Fenoldopam is an agonist for peripheral D1 receptors and binds with moderate affinity to α2 adrenergic receptors; it has no significant affinity for D2 receptors or α1 or β adrenergic receptors. Fenoldopam is a racemic mixture; the R-isomer is the active component. It dilates a variety of blood vessels, including coronary arteries, afferent and efferent arterioles in the kidney, and mesenteric arteries. Less than 6% of an orally administered dose is absorbed because of extensive first-pass metabolism. The elimination t1/2 of intravenously infused fenoldopam is ~10 min. Adverse effects are related to the vasodilation and include headache, flushing, dizziness, and tachycardia or bradycardia.
Dopexamine (DOPACARD) is a synthetic analog related to DA with intrinsic activity at dopamine D1 and D2 receptors as well as at β2 receptors; it may have other effects such as inhibition of catecholamine uptake. It has favorable hemodynamic actions in patients with severe congestive heart failure, sepsis, and shock. In patients with low cardiac output, dopexamine infusion significantly increases stroke volume with a decrease in systemic vascular resistance. Tachycardia and hypotension can occur, but usually only at high infusion rates. Dopexamine is not currently available in the U.S.
β ADRENERGIC RECEPTOR AGONISTS
β agonists play a major role only in the treatment of bronchoconstriction in patients with asthma (reversible airway obstruction) or chronic obstructive pulmonary disease (COPD). Minor uses include management of preterm labor, treatment of complete heart block in shock, and short-term treatment of cardiac decompensation after surgery or in patients with congestive heart failure or myocardial infarction. β Receptor agonists may be used to stimulate the rate and force of cardiac contraction. The chronotropic effect is useful in the emergency treatment of arrhythmias such as torsade de pointes, bradycardia, or heart block (see Chapter 29).
ISOPROTERENOL
Isoproterenol (see Table 12–1) is a potent, nonselective β receptor agonist with very low affinity for α receptors. Consequently, isoproterenol has powerful effects on all β receptors and almost no action at α receptors.
PHARMACOLOGICAL ACTIONS. The major cardiovascular effects of isoproterenol (compared with EPI and NE) are illustrated in Figure 12–2. IV infusion of isoproterenol lowers peripheral vascular resistance, primarily in skeletal muscle but also in renal and mesenteric vascular beds. Diastolic blood pressure falls. Systolic blood pressure may remain unchanged or rise, although mean arterial pressure typically falls. Cardiac output is increased because of the positive inotropic and chronotropic effects of the drug in the face of diminished peripheral vascular resistance. The cardiac effects of isoproterenol may lead to palpitations, sinus tachycardia, and more serious arrhythmias.
Isoproterenol relaxes almost all varieties of smooth muscle when the tone is high, but this action is most pronounced on bronchial and GI smooth muscle. It prevents or relieves bronchoconstriction. Its effect in asthma may be due in part to an additional action to inhibit antigen-induced release of histamine and other mediators of inflammation; this action is shared by β2-selective stimulants.
ABSORPTION, FATE, AND EXCRETION. Isoproterenol is readily absorbed when given parenterally or as an aerosol. It is metabolized primarily in the liver and other tissues by COMT. Isoproterenol is a relatively poor substrate for MAO and is not taken up by sympathetic neurons to the same extent as are EPI and NE. The duration of action of isoproterenol therefore may be longer than that of EPI, but it still is brief.
TOXICITY AND ADVERSE EFFECTS. Palpitations, tachycardia, headache, and flushing are common. Cardiac ischemia and arrhythmias may occur, particularly in patients with underlying coronary artery disease.
THERAPEUTIC USES. Isoproterenol (ISUPREL, others) may be used in emergencies to stimulate heart rate in patients with bradycardia or heart block, particularly in anticipation of inserting an artificial cardiac pacemaker or in patients with the ventricular arrhythmia torsade de pointes. In disorders such as asthma and shock, isoproterenol largely has been replaced by other sympathomimetic drugs (see below and Chapter 36).
DOBUTAMINE
The pharmacological effects of dobutamine (see Table 12–1) are due to direct interactions with α and β receptors and are complex.
Dobutamine possesses a center of asymmetry; both enantiomeric forms are present in the racemic mixture used clinically. The (–) isomer of dobutamine is a potent agonist at α1 receptors and is capable of causing marked pressor responses; (+)-dobutamine is a potent α1 receptor antagonist, which can block the effects of (–)-dobutamine. Both isomers are full agonists at β receptors, but the (+) isomer is a more potent β receptor agonist than the (–) isomer (~10-fold).
CARDIOVASCULAR EFFECTS. The cardiovascular effects of racemic dobutamine are a composite of the pharmacological properties of the (–) and (+) stereoisomers. Dobutamine has relatively more prominent inotropic than chronotropic effects on the heart compared to isoproterenol. Although not completely understood, this useful selectivity may arise because peripheral resistance is relatively unchanged. Alternatively, cardiac α1 receptors may contribute to the inotropic effect. At equivalent inotropic doses, dobutamine enhances automaticity of the sinus node to a lesser extent than does isoproterenol; however, enhancement of atrioventricular and intraventricular conduction is similar for both drugs.
ADVERSE EFFECTS. Blood pressure and heart rate may increase significantly during dobutamine administration requiring reduction of infusion rate. Patients with a history of hypertension may exhibit an exaggerated pressor response more frequently. Because dobutamine facilitates AV conduction, patients with atrial fibrillation are at risk of marked increases in ventricular response rates; digoxin or other measures may be required to prevent this from occurring. Some patients may develop ventricular ectopic activity. As with any inotropic agent, dobutamine may increase the size of a myocardial infarct by increasing myocardial O2 demand. The efficacy of dobutamine over a period of more than a few days is uncertain; there is evidence for the development of tolerance.
THERAPEUTIC USES. Dobutamine (DOBUTREX) is indicated for the short-term treatment of cardiac decompensation that may occur after cardiac surgery or in patients with congestive heart failure or acute myocardial infarction. Dobutamine increases cardiac output and stroke volume in such patients, usually without a marked increase in heart rate. Alterations in blood pressure or peripheral resistance usually are minor. An infusion of dobutamine in combination with echocardiography is useful in the noninvasive assessment of patients with coronary artery disease.
Dobutamine has a t1/2 ~2 min; the onset of effect is rapid, steady-state concentrations are achieved within 10 min, and major metabolites are conjugates of dobutamine and 3-O-methyldobutamine. The rate of infusion required to increase cardiac output is 2.5-10 μg/kg/min; higher infusion rates occasionally are required. The rate and duration of the infusion are determined by the clinical and hemodynamic responses of the patient.
β2-SELECTIVE ADRENERGIC RECEPTOR AGONISTS
In the treatment of asthma or COPD, drugs with preferential affinity for β2 receptors compared with β1 receptors have been developed. The selectivity, however, is not absolute, and is lost at high concentrations of these drugs. Moreover up to 40% of the β receptors in the human heart are β2 receptors, activation of which can cause cardiac stimulation. A useful strategy to enhance preferential activation of pulmonary β2 receptors is the administration by inhalation of small doses of the drug in aerosol form. This approach typically leads to effective activation of β2 receptors in the bronchi but very low systemic drug concentrations. Consequently, there is less potential to activate cardiac β1 or β2 receptors or to stimulate β2 receptors in skeletal muscle, which can cause tremor and thereby limit oral therapy.
Administration of β agonists by aerosol (see Chapter 36) typically leads to a very rapid therapeutic response, generally within minutes, although some agonists such as salmeterol have a delayed onset of action. Only ~10% of an inhaled dose actually enters the lungs; much of the remainder is swallowed and ultimately may be absorbed.
In the treatment of asthma and COPD, β receptor agonists are used to activate pulmonary receptors that relax bronchial smooth muscle and decrease airway resistance. β receptor agonists also may suppress the release of leukotrienes and histamine from mast cells in lung tissue, enhance mucociliary function, and decrease microvascular permeability.
SHORT-ACTING β2 ADRENERGIC AGONISTS
METAPROTERENOL. Metaproterenol (called orciprenaline in Europe), along with terbutaline and fenoterol, belongs to the structural class of resorcinol bronchodilators that have hydroxyl groups at positions 3 and 5 of the phenyl ring (rather than at positions 3 and 4 as in catechols) (see Table 12–1). Consequently, metaproterenol is resistant to methylation by COMT. It is excreted primarily as glucuronic acid conjugates. Metaproterenol is considered to be β2-selective, although it probably is less selective than albuterol or terbutaline and hence is more prone to cause cardiac stimulation.
Effects occur within minutes of inhalation and persist for several hours. After oral administration, onset of action is slower, but effects last 3-4 h. Metaproterenol is used for the long-term treatment of obstructive airway diseases, asthma, and for treatment of acute bronchospasm. Side effects are similar to the short- and intermediate-acting sympathomimetic bronchodilators.
ALBUTEROL. Albuterol (VENTOLIN-HFA, PROVENTIL-HFA, others; see Table 12–1) is a selective β2 receptor agonist with pharmacological properties and therapeutic indications similar to those of terbutaline. It is administered either by inhalation or orally for the symptomatic relief of bronchospasm.
When administered by inhalation, it produces significant bronchodilation within 15 min, and effects persist for 3-4 h. The cardiovascular effects of albuterol are considerably weaker than those of isoproterenol when doses that produce comparable bronchodilation are administered by inhalation. Oral albuterol has the potential to delay preterm labor. Although rare, CNS and respiratory side effects are sometimes observed.
LEVALBUTEROL. Levalbuterol (XOPENEX
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


