GENERAL FEATURES
Nomenclature
Acute leukemia arising from myeloid cells has been variously referred to as myeloblastic, myelocytic, and myeloid, with little, if any, clarification as to the meaning implied by these different terms. In this chapter, the overall designation “myeloid” is used for acute leukemias arising from myeloid cells, regardless of the maturity of the phenotype.
The term myeloid can refer specifically to neutrophils or granulocytes, or can refer more generally to all hematopoietic cells primarily generated in the bone marrow, regardless of lineage. This chapter uses the term myeloid in a general sense, to include all hematopoietic lineages arising from the myeloid stem cell, including neutrophils, eosinophils, basophils, monocytes, mast cells, erythroid cells, and megakaryocytes.
Given the wide variety of cell types arising from myeloid stem cells, it is not surprising that myeloid malignancies encompass a broad range of phenotypes, including multilineal and ambiguous phenotypes.
Definition and Classification
Acute myeloid leukemia (AML) is a malignant clonal proliferation of myeloid cells, usually expressing an immature immunophenotype and one or more recurrent genotypes (
1,
2,
3,
4,
5,
6,
7). AML can be classified according to morphology, cytochemistry, immunophenotype, and genotype. The most recent classification of AML is that formulated by the World Health Organization (WHO) in 2008 (see
Appendix A for WHO 2008 classifications) and is based primarily on immunophenotype and genotype. The earlier French-American-British (FAB) classification is still widely used and is based primarily on morphology and cytochemistry.
According to current practice, AML is diagnosed at a peripheral blood or bone marrow blast percentage of 20% or more. Cases presenting primarily as tissue infiltrates or masses with are designated granulocytic sarcoma.
Discussed below are subtypes of AML according to both WHO and FAB criteria, as well as other subtypes that have clinicopathologic significance but are not included in either classification.
Etiology
AML arises from many different conditions, discussed further in
Chapter 17. Some factors are constitutional, or inherited, and some are somatic, or acquired.
AML occurs de novo and as a terminal event in myeloproliferative disorders and myelodysplastic syndrome, and, uncommonly, as a transformation of preexisting lymphoma.
Clearly, in these and other cases, acquisition of a genetic anomaly is necessary but not sufficient for the development of acute leukemia. Combinations of risk factors are common, suggesting that multiple factors are required to initiate and sustain clonal expansion.
Clinical Course and Prognosis
AML pursues a generally downward course, complicated by bone marrow failure caused by the leukemia itself and by antileukemia therapy. Combination chemotherapy induces remission in more than one half of patients but is followed eventually by relapse in most. Stem cell transplantation offers the best chance of long-term remission or cure.
Prognosis is primarily linked to genotype and possibly also to age (
8). Approximately 25% to 30% of AML cases carry recurrent genetic abnormalities, 20% show multilineage dysplasia, less than 1% are biphenotypic, and approximately 50% lack these features. Relatively good prognosis is found in AML with t(8;21), t(15;17), and inv(16). Relatively poor prognosis is seen in cases with multiple and complex genetic abnormalities and deletions of 5’5q and 7’7q. Intermediate prognosis is found in other cases, including those with chromosome 11q23 and’or
MLL anomalies.
Spontaneous remission occasionally occurs in AML and may be transient, with eventual relapse, or permanent, with complete disappearance of leukemia (
9,
10,
11,
12,
13,
14,
15,
16,
17,
18).
In infants, spontaneous remission is associated with congenital leukemia, monocytic differentiation, and skin involvement. It may occur spontaneously, with no other disease or therapy.
In older children and adults, spontaneous remission is more often associated with conditions affecting immunity and granulocyte production. Conditions associated with remission include serious infection and sepsis, antibiotic therapy, hypergammaglobulinemia, immunoglobulin therapy, blood transfusion, and therapy with corticosteroid, granulocyte colony-stimulating factor, and valproic acid. Withdrawal of G-CSF and valproic acid led to remission. Administration of triptorelin, a gonadotropin-releasing hormone agonist, has also been reported to induce remission.
Peripheral Blood Findings and Blast Morphology
The peripheral blood usually shows anemia and thrombocytopenia. The red blood cells may show aniso- and poikilocytosis. Platelets may be of abnormal size and show hypogranularity. The white blood cell count and blast percentage are highly variable. A peripheral blood blast percentage of 20% warrants a diagnosis of acute leukemia, even if the bone marrow blast percentage is lower (
19). In cases of pancytopenia, the peripheral blood smear should be carefully searched for blasts.
Dysplastic neutrophils, monocytes, and nucleated red blood cells often appear in the peripheral blood in AML and are clues to the diagnosis. The peripheral blood may show other hematologic abnormalities, such as autoimmune hemolytic anemia (
20).
Blast morphology is highly variable in AML and is discussed in greater detail with the morphologic subtypes. In general, blasts range from approximately 15 to 30 μm in diameter. The nucleus is round to elongated, often showing folds or other nuclear membrane irregularities. The chromatin is finely divided and nucleoli are usually visible.
Blast cytoplasm ranges from scant to abundant and usually contains small primary (azurophilic or reddish) granules (
21,
22,
23,
24). In approximately 20% of cases, the primary granules coalesce into needle-like Auer rods, irregular Auer bodies, or Chediak-Higashi-like inclusions. The latter seem to be associated with CD2 expression. Multiple intracellular Auer rods are a feature of acute promyelocytic leukemia.
Blast size occasionally provides a clue to the karyotype. Small blast size and hand-mirror morphology have been described in AML with trisomy 13 (
25). Huge blast size and bizarre morphology have been reported in AML with polyploidy (
26,
27,
28,
29,
30,
31,
32,
33).
Blasts occasionally show hemophagocytosis, especially in AML with t(8;16)(p11;p13) and other translocations of chromosome 8p11 involving
MYST3, and in AML with t(16;21)(p11;q22) and
FUS’ERG fusion (
34,
35,
36,
37,
38,
39).
Bone Marrow
Bone marrow aspirate smears typically show 20% or more blasts, admixed with other hematopoietic cells, lymphocytes, and plasma cells. The myeloid:erythroid ratio may be unevaluable because of blast infiltration. Differentiated hematopoietic cells often show dysplastic features, especially in AML arising from myelotoxic exposure or preexisting hematopoietic disease.
Histologic sections of the marrow show hypercellularity for site and age (
40). Hypoplastic or hypocellular AML refers to cases with cellularity of 30% or less (
41). Use of this arbitrary criterion does not take into account normal siteand age-specific cellularity and does not appear to define a clinicopathologic entity (
42).
Other bone marrow findings in AML include reactive plasmacytosis, granulomas, fibrosis, osteosclerosis, osteoporosis, and hyperplasia of precursor B cells (
43).
Other diseases reported with AML include sarcoidosis (
44,
45) and clonal histiocytic, B-cell, plasma cell, and T-cell disorders (
46,
47,
48,
49,
50,
51,
52,
53). Coexistent neoplasms may be either clonally related or unrelated to the AML.
Cytochemistry
Cytochemistry is less important than it once was in the diagnosis of AML (
54,
55,
56). In brief, granulocytic differentiation is revealed by cytoplasmic positivity for Sudan black B and myeloperoxidase (MPO) and, in more differentiated blasts, chloroacetate esterase. Monocytic differentiation is revealed by cytoplasmic positivity for nonspecific esterase (NSE), inhibitable by sodium fluoride. AML with t(8;21) usually shows myeloid differentiation and thus has a higher number of MPO-positive blasts. A high percentage of MPO-positive blasts correlates with morphologic subtype but is not an independent prognostic factor.
Immunophenotyping by Flow Cytometry
Immunophenotyping by flow cytometry is essential for the accurate diagnosis and classification of acute leukemia and for monitoring minimal residual disease (
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72).
AML blasts commonly express surface antigens CD13, CD14, CD15, CD33, and CD117, and intracellular myeloperoxidase (MPO). CD45 is expressed in more than 95% of cases. HLA-DR is expressed in 85% of cases, and CD34 in 60% of cases. CD13 and MPO, the most sensitive myeloid-specific antigens, are each expressed in 75% to 95% of cases. CD33 expression is seen in 65% to 95% of cases. CD117 expression, seen in approximately 70% of cases, is virtually specific for a diagnosis of AML.
Compared with their normal counterparts, leukemic myeloblasts show cross-lineage and asynchronous antigen expression in almost 90% of cases. CD4 is expressed in 60% of cases; CD7, CD19, and CD20 in 10% to 20%; and CD2, CD3, CD5, and CD10 in 5% to 10%. CD2 expression is characteristic of acute promyelocytic leukemia and acute myelomonocytic leukemia with eosinophilia. Coexpression of CD7 with the early hematopoietic antigens CD34, CD117, and terminal deoxynucleotidyl transferase (TdT) reproduces a stem cell immunophenotype and augurs a poor prognosis. Expression of nonmyeloid markers in AML is helpful in distinguishing AML from regenerating, nonclonal blasts.
CD79 alpha is expressed not only in B-cell neoplasms, but also in approximately 90% of acute promyelocytic leukemia cases and 5% of other AML cases.
Other antigens that may be expressed in AML are CD11b, CD11c, CD14, CD15, CD34, CD56, and TdT. TdT expression is found in about 50% of AML. CD14 and CD15 expression is typically found in cases with monocytic differentiation. CD56, expressed in 24% of cases, appears to predict a poor prognosis.
Immunophenotype is not necessarily stable in AML. Immunophenotypic changes occur in more than 90% of AML cases over time. Relapses may show gain of CD13, CD33, and CD34 expression and’or loss of CD14, CD19, and CD56 expression. Immunophenotype switching from myeloid to lymphoid immunophenotype and vice versa (see later). Immunophenotype is further discussed with each subtype of AML.
Immunophenotyping by Histochemistry
Immunophenotyping by immunohistochemistry may be used for classification of AML (
73,
74,
75,
76,
77). This method is especially useful for the diagnosis of extramedullary disease, when no tissue may have been submitted for immunophenotyping by flow cytometry.
Immunohistochemistry shows some similarities and differences from immunophenotyping by flow cytometry. CD33 and CD34 detection are similar. For CD15 and CD117 detection, flow cytometry is more sensitive than histochemistry. For MPO detection, histochemistry is more sensitive.
Numerous antibodies are available for immunohistochemistry. Using a panel of CD3, CD20, CD34, CD43, CD68, CD79a, HLA-DR, myeloperoxidase (MPO), and terminal deoxynucleotidyl transferase, more than 95% of acute leukemias can be correctly identified as either acute lymphoblastic or acute myeloid leukemia. AML blasts have shown MPO positivity in about 85% of AML cases, FAB subtypes of M1-M4 and M6. Cases with monocytic differentiation usually stain for CD68. Cases of acute erythoid leukemia consistently stain for hemoglobin. Some, but not all, cases of acute megakaryocytic leukemia stain with factor VIII.
GENETIC ANALYSIS
Genetic analysis is invaluable in AML for assessing clonality and detecting prognostically important anomalies (
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97,
98,
99). Genotype is the single most important predictor of prognosis. Clonal karyotypic abnormalities are found in approximately 90% of childhood and 60% of adult cases. The figure is higher when more sensitive molecular methods are used. Molecular methods are used to detect clonal abnormalities of the mutational status of
FLT3, NPM1, FLT3, CEBPA, and
MLL, which are found in karyotypically normal AML cases.
The most common karyotypic findings are loss of a sex chromosome; deletions of 5, 5q, 7, and 7q; trisomy 8; rearrangements of 3q21-26, 11q23, and 12q13; translocations (8;21), (1;22), (6;9), (8;21), (9;22), and (15;17); and inversion of chromosome 16. Rare cases of AML are multiclonal (
75).
Nonspecific rearrangements of the immunoglobulin heavy chain gene and T-cell receptor genes are found in approximately 35% to 45% of cases expressing B-cell
antigens, and 10% of cases expressing T-cell antigens. Rearrangements are rarely found in cases lacking these antigens and are not predictive of genotype or prognosis. A rare biclonal case of AML has been reported.
AML subtypes with clinically significant genotypes are described below in greater detail.
Differential Diagnosis
The differential diagnosis of AML includes a wide array of nonneoplastic and neoplastic conditions (
Table 18.2) (
100,
101,
102,
103,
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128,
129,
130,
131,
132,
133).
Nonneoplastic conditions resembling AML are a heterogeneous group. Many of these disorders have been reported to coexist with myelodysplastic syndrome (MDS) and AML. When occurring in MDS, they may result in a mistaken or premature diagnosis of AML.
Leukemoid reactions may consist predominantly of blasts, promyelocytes, or monocytes and thus be mistaken for AML or acute promyelocytic or monocytic leukemia.
Infectious monocytosis may be misdiagnosed in patients with sore throat and atypical circulating cells who have acute monocytic leukemia. It should be kept in mind that infectious mononucleosis and AML have been reported to occur together.
Therapy with granulocyte colony-stimulating factor therapy may cause a transient increase in peripheral blood and bone marrow monocytes or blasts.
Florid folate- or vitamin B12-related megaloblastic change is characterized by pancytopenia, bone marrow hyperplasia, increased early erythroid precursors with fine, blast-like chromatin, and numerous mitotic figures arrested in metaphase. These findings are easily misinterpreted as acute leukemia if histologic sections are examined without corresponding bone marrow aspirate smears.
Precursor B-cell hyperplasia has been reported in AML and must be distinguished from a myeloid-to-lymphoid immunophenotype switch and from therapy-related acute lymphoblastic leukemia.
Neoplastic conditions resembling AML include malignant lymphoma, malignant histiocytosis, and nonhematolymphoid tumors, especially neuroblastoma and embryonal rhabdomyosarcoma. These disorders may present with a leukemic phase and’or massive bone marrow involvement. It should be kept in mind that AML has been reported with other hematolymphoid neoplasms and with neuroblastoma.
MORPHOLOGIC AND IMMUNOPHENOTYPIC SUBTYPES
The WHO term “AML not otherwise classified” is a broad definition. It includes cases which can be grouped by morphology and immunophenotype, determined primarily by predominant lineage. Combinations of lineages, such as granulocytic and monocytic or megakaryocytic and erythroid, also occur. Such cases are termed “acute leukemias of ambiguous lineage.” Rare cases cannot be classified any further than acute undifferentiated or stem cell leukemia. Acute leukemia may preserve a stable genotype, but change phenotype, in a “phenotype switch.”
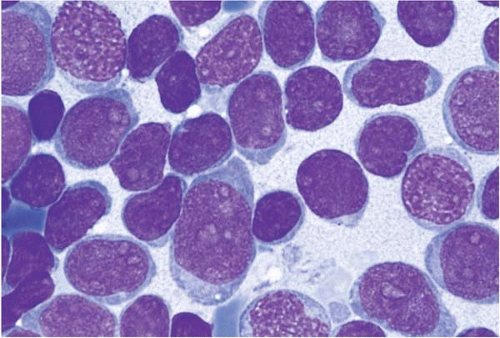
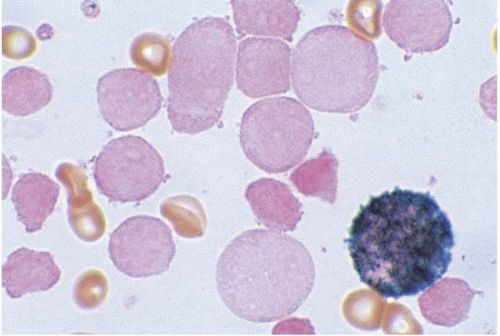
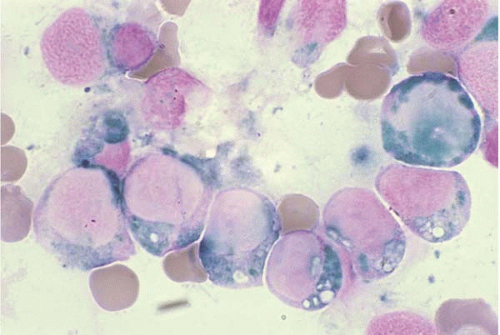
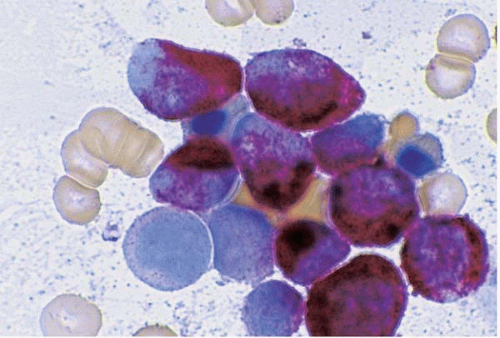
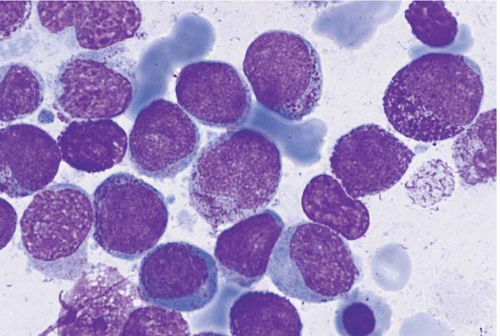
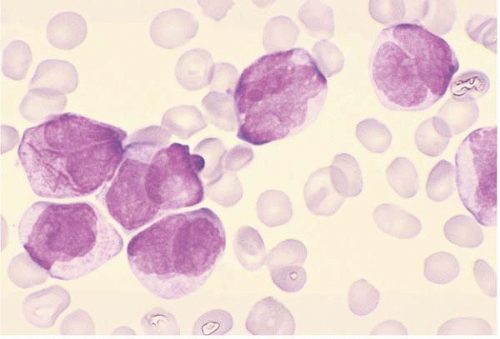
Acute Myeloid Leukemia with Minimal Differentiation (FAB M0)
AML with minimal differentiation (FAB M0) accounts for approximately 5% of AML cases (
Figs. 18.1 and
18.2) (
134,
135,
136,
137,
138,
139,
140,
141,
142,
143). It occurs primarily in children younger than 3 years old and adults older than 60 years old. The peripheral white blood cell count is variable.
The majority of blasts lack cytoplasmic granules, Auer rods, and cytochemical markers of myeloid differentiation.
Other hematopoietic cells may show myelodysplastic changes. Immunophenotyping consistently shows expression of myeloperoxidase. CD13 and CD33 are variably expressed. The stem cell markers CD7, CD34, HLA-DR, and TdT are frequently expressed.
The genotype resembles that found in myelotoxin- and therapy-related AML. Cytogenetic anomalies are found in more than 70% of cases, particularly +8, +11, +13, and +14, 5’5q-, 7’7q-, t(12)(p), and t(8;21) and other translocations involving RUNX1 on chromosome 21q22.3. Nonspecific rearrangements of immunoglobulin and T-cell receptor genes may also be present. Cases of FAB M0 expressing CD7 and CD56 may be viewed as acute myeloid’NK cell leukemia, and have a notably worse prognosis (see below).
The prognosis is poor.
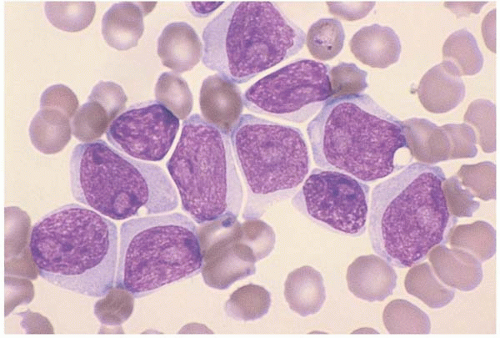
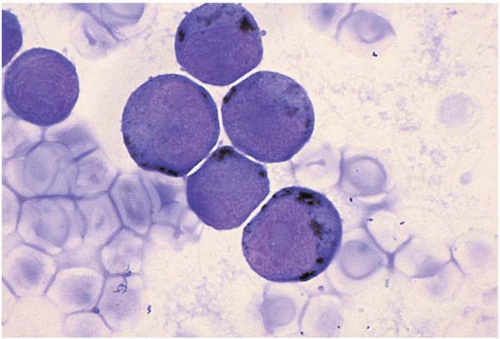
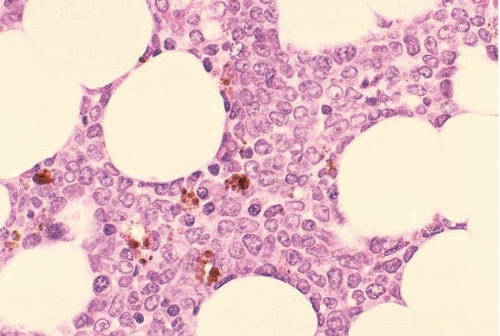
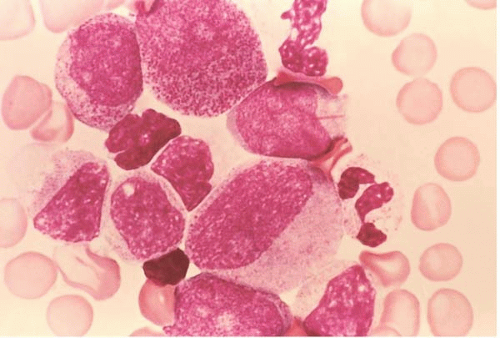
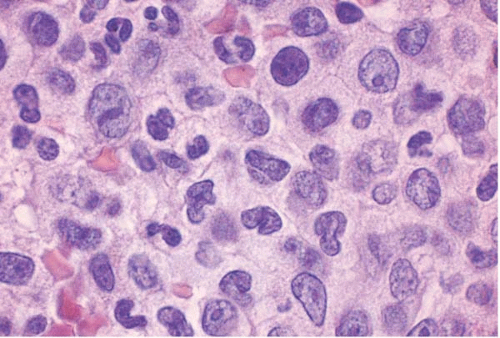
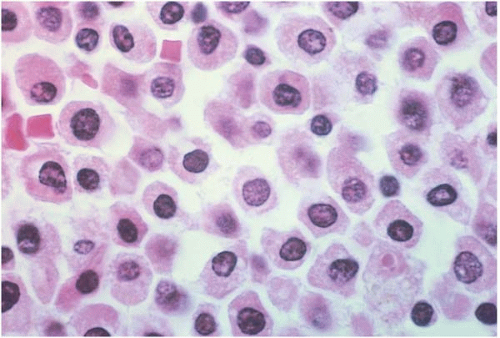
Acute Myeloid Leukemia without Maturation (FAB M1)
AML without maturation is the most common morphologic subtype of AML (
Figs. 18.3,
18.4,
18.5and
18.6) (
144,
145,
146,
147). The blasts typically contain primary granules and are positive for Sudan black B and’or myeloperoxidase. Auer rods are often present. An unusual case has been reported, showing bundles (faggots) of Auer rods in blasts, mature segmented neutrophils, and metamyelocytes. Faggot cells are typically found in the blasts of acute promyelocytic leukemia (FAB M3), and even then, they not usually seen in more mature granulocytes.
Immunophenotyping shows consistent expression of CD13, CD33, CD34, and HLA-DR, and variable expression of CD7 and TdT.
Genetic studies show one or more abnormalities in approximately 50% of cases. These include +8, +13, +14, +11, t(9;22), 11q23 anomalies, and MLL rearrangement. Translocation (8;21) is uncommon.
The prognosis is intermediate.
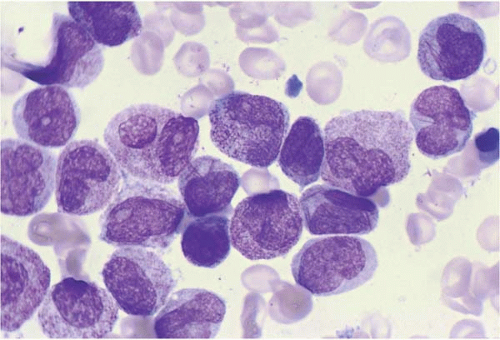
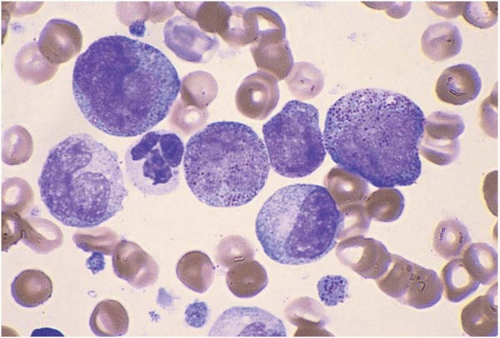
Acute Myeloid Leukemia with Maturation (FAB M2)
AML with maturation shows a mixture of blasts, dysplastic neutrophil precursors, and dysplastic segmented neutrophils (
Figs. 18.7,
18.8,
18.9and
18.10) (
148,
149,
150,
151,
152,
153,
154). Auer rods are often present in blasts and may even be seen in segmented neutrophils and bands. Some cases show eosinophilic, basophilic, and’or mast cell differentiation; these are discussed
separately below. Extramedullary disease is relatively frequent in cases with t(8;21).
Immunophenotyping may show expression of CD13, CD15, CD19, CD33, and CD34. CD79a may be expressed, a finding associated with t(8;21). CD79a is characteristic of AML M2 with t(8;21) and should not be construed as evidence of biphenotypism. CD7 and CD56 may be expressed.
Approximately 50% of cases show an abnormal karyotype. Recurrent anomalies include +8, +13, +14, and +11. About 10% to 15% of cases show a distinctive anomaly, t(8;21)(q22;q23), in which RUNX1T1 (previously known as ETO) on chromosome 8q22 is fused to RUNX1 on chromosome 21q22.3. Such cases tend not to show CD13 expression. Some cases show chromosome 11q23 translocation; these cases tend not to show dysgranulopoiesis or Auer rods.
The prognosis is relatively good.
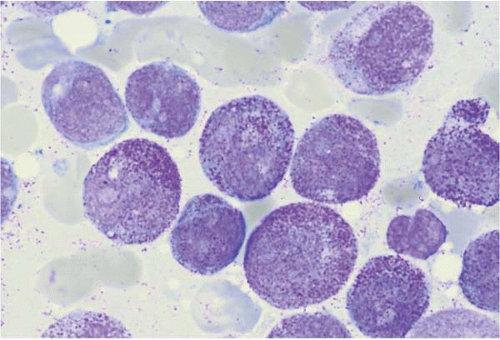
Acute Promyelocytic Leukemia (FAB M3)
APL accounts for approximately 10% to 15% of AML cases (
Figs. 18.11,
18.12,
18.13and
18.14) (
155,
156,
157,
158,
159,
160,
161,
162,
163,
164,
165,
166,
167,
168,
169,
170,
171,
172,
173,
174,
175,
176,
177). Its clinical course is marked by life-threatening disseminated fibrinolysis, owing to the release of granule contents from neoplastic cells. The initial mortality rate is high due to the coagulopathy.
Laboratory studies may show leukopenia and few blasts, and the diagnosis may therefore be missed initially. Careful
study of the peripheral blood smear may reveal blasts in such cases. Abnormal coagulation studies may be present.
APL is morphologically variable. The hypergranular blast predominates in most cases. The nucleus is large and lobated, and the cytoplasm abundant and heavily granulated. Auer rods are typically (but not always) present and may be multiple within a single cell, forming bundles or faggots.
The smaller microgranular blast predominates in 15% to 20% of cases (FAB M3v). The nucleus is bilobed or folded and the cytoplasm is moderately abundant, with fine dust-like or submicroscopic granules. The cytoplasm may appear hypo- or agranular with Wright stain because of the small granule size. The lobated nucleus and apparently agranular cytoplasm may prompt an erroneous diagnosis of acute monocytic leukemia.
Basophilic APL is a rare variant associated with hyperhistaminemia. The blasts show scant, basophilic cytoplasm with large, deep violet granules and vacuoles. The granules may be scarce. Like basophil granules, they stain metachromatic (pink) with toluidine blue stain.
Some cases of APL show predominantly promyelocyte-like cells but few blasts, thus resembling AML with maturation (FAB M2). In some cases, the predominant blast is a promyelocyte-like cell with innumerable large, dark granules but no Auer rods. In other cases, blasts with a few small primary granules predominate, thus resembling AML without maturation. Large, abnormal granules may resemble Chediak-Higashi anomaly. Histologic sections of APL show marrow replacement by cells with reniform or lobated nuclei surrounded by abundant, faintly eosinophilic cytoplasm, creating a distinctive “fried-egg” appearance. Marked fibrosis has been reported.
Cytochemical staining of hypergranular blasts shows strong reactions for Sudan black B, myeloperoxidase (MPO), and chloroacetate esterase, and, in some cases, fluoride-sensitive nonspecific esterase (NSE). Microgranular blasts show weaker reactions. Basophilic APL shows weaker MPO staining and may be NSE-positive.
Immunophenotyping results correspond to the degree of morphologic maturation. Hypergranular blasts show a mature myeloid phenotype, with expression of CD13 and CD33 but lacking expression of CD2, CD4, CD7, CD14, CD15, CD19, CD34, and HLA-DR. Microgranular blasts show a more immature phenotype, with coexpression of CD2 and CD19, CD34, and, in some cases, CD56 and HLA-DR. Rare cases show an even more primitive phenotype, with expression of CD2, CD13, CD33, CD34, CD117, and TdT. Although APL may show expression of either CD34 or HLA-DR, it does not characteristically show expression of both.
Genetic studies show t(15;17) in more than 90%, but not all, APL cases. APL and AML with t(15;17) are not identical. Some cases of APL show promyelocytic morphology but lack t(15;17). Others show t(15;17) but lack promyelocytic morphology. The presence or absence of t(15;17), not morphology, is the determining prognostic and therapeutic factor.


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
