INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should understand:
The pathogenic role of mutations in genes encoding transcription factors and signaling molecules in acute leukemia.
The diagnostic and prognostic role of immunophenotyping, cytogenetics, and molecular genetics in acute leukemia.
The general approach to treatment of patients with acute leukemia and the associated short-term complications.
The unique role of “differentiating” agents in the treatment of acute promyelocytic leukemia.
PATHOGENESIS OF ACUTE LEUKEMIA
Acute leukemias and related disorders are aggressive neoplasms caused by acquired somatic mutations in early hematopoietic progenitors. The most obvious pathologic feature in the acute leukemias is the accumulation of undifferentiated blasts in the marrow and other tissues, indicating that, unlike the myeloproliferative disorders, acute leukemias have defects that block or significantly retard differentiation. We now know that specific subtypes of acute leukemia are often associated with mutations that alter the function of transcription factors that are required for normal differentiation of hematopoietic progenitors (Table 21-1). Sometimes these mutations consist of chromosomal rearrangements that create chimeric fusion genes, in which one or both partners encode a transcription factor; in other cases, the pathogenic mutations are more subtle point mutations or deletions. In most instances, the net result of the mutations is to decrease the function of a transcription factor that is required for the differentiation of cells of one or another of the hematopoietic lineages. An exception is mutations in the NOTCH1 gene, which increase the transcriptional activity of the NOTCH1 protein.
| Mutations | Effects | Normal Function of Aff ected Gene(s) | Associated Acute Leukemia |
|---|---|---|---|
| PML-RARαfusion gene [t(15;17)] | Decreased RARα function | RARα: required for granulopoiesis | Acute promyelocytic leukemia |
| C/EBPA point mutations | Decreased C/EBPα function | Required for granulopoiesis | Acute myeloid leukemia |
| PAX5, E2A, and EBF deletions | Decreased PAX5, E2A, and EBF function | Required for early stages of B-cell development | B-cell acute lymphoblastic leukemia/lymphoblastic lymphoma |
| NOTCH1 point mutations | Increased NOTCH1 function | Required for early stages of T-cell development | T-cell acute lymphoblastic leukemia/lymphoblastic lymphoma |
The expression of NOTCH1 genes bearing gain-of-function mutations in mouse hematopoietic stem cells causes the rapid development of T-cell acute lymphoblastic leukemia/lymphoma (T-ALL), the disease that is specifically associated with NOTCH1 mutations in man, proving that these mutations are leukemogenic. However, similar experiments performed with other transcription factor genes bearing leukemia-associated mutations that decrease or interfere with a normal transcriptional activity usually fail to produce acute leukemia or do so only after very long periods of time. In fact, when expressed in hematopoietic stem cells, some mutated transcription factors cause bone marrow failure, suggesting that their primary effect is to block differentiation rather than to cause proliferation. Further evidence that transcription factor mutations are not sufficient to cause acute leukemia has been gleaned from retrospective studies using blood samples (so-called Guthrie cards) that are obtained routinely from newborns. Analyses of these samples by using sensitive polymerase chain reaction (PCR)-based assays have shown that certain transcription factor mutations were present at birth in children who developed acute leukemia as much as 10 to 12 years later. Thus, even acute leukemia may have a long prodrome, during which “pre-leukemic” clones bearing an initial mutation in a transcription factor gene must acquire other somatic mutations for full-blown disease to appear. These insights have provided support for a model in which the development of acute leukemia requires at least two complementary events, a class 1 mutation that drives proliferation and a class 2 mutation in a transcription factor that arrests differentiation (Fig. 21-1).
FIGURE 21-1
Types of mutations in hematologic malignancies. Most myeloproliferative disorders (discussed in Chapter 20) have activating class 1 mutations in tyrosine kinases that stimulate growth factor–independent proliferation but leave differentiation relatively intact. Most acute leukemias have mutations that interfere with or alter the activity of one or more transcription factors that are required for normal differentiation. These mutations (class 2), when introduced into the hematopoietic stem cells of mice, cause a maturation arrest but are not sufficient to cause acute leukemia. It is hypothesized that acute leukemias arise from transformed hematopoietic progenitors that acquire both class 1 and class 2 mutations, which collaborate to drive proliferation and block differentiation.
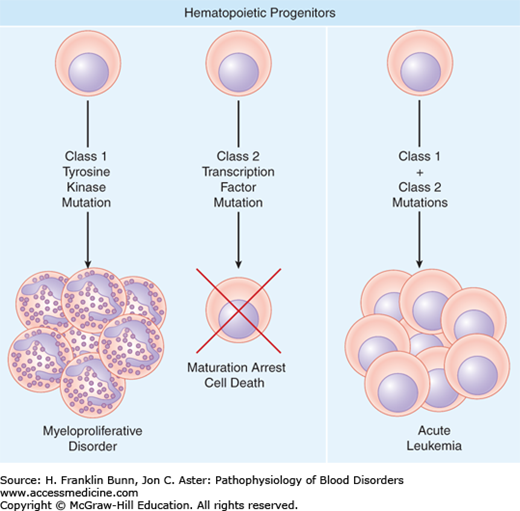
The class 1 mutations in at least some acute leukemias appear to be gain-of-function mutations in tyrosine kinases, similar to the mutations that are prevalent in the myeloproliferative disorders (Chapter 20). It has long been recognized that a subset of acute lymphoblastic leukemia/lymphoma of B-cell origin (B-ALL) is associated with the presence of the Philadelphia chromosome and a BCR-ABL fusion gene. At the cytogenetic level, this is the same lesion found in chronic myelogenous leukemia (CML), a classic myeloproliferative disorder that, if untreated, usually transforms to blast crisis and a picture identical to that of acute leukemia. At a molecular level, it turns out that the form of BCR-ABL protein in B-ALL is usually 190 kDa in size, slightly smaller than the 210 kDa form usually found in CML. However, the 190 kDa form of BCR-ABL activates the same pathways (RAS, JAK/STAT, and PI-3 kinase/AKT) as the 210 kDa form, differing mainly in having a stronger tyrosine kinase activity. When expressed in the hematopoietic stem cells of mice, both the 210 kDa and 190 kDa forms of BCR-ABL produce a myeloproliferative disorder similar to CML.
These observations suggested that BCR-ABL–positive ALLs and B-cell blast crises arising in CML might share the same complementary class 2 mutation in some critical transcription factor. Recently, this prediction has been confirmed with the detection of mutations in a gene called Ikaros in up to 90% of these two forms of acute leukemia. Ikaros is a transcription factor that regulates early stages of lymphocyte development. The Ikaros mutations result in expression of truncated forms of the protein that interfere with its normal function and thereby prevent the normal differentiation of early lymphoid progenitors. Compellingly, the Ikaros mutations found in the blast crisis stage are absent from the preceding stable phase of CML, linking them directly to the transformation of the myeloproliferative disorder to acute leukemia. It is hypothesized that other transformations, such as the progression of myelodysplastic syndromes to acute myeloid leukemia (AML), are also due to as-yet-unknown acquired class 2 mutations that lead to the loss of some critical transcription factor activity that is required for differentiation.
The mutations that conspire to produce acute leukemia are still being discovered, but it is clear that many different combinations of class 1 and class 2 mutations may be involved, including some that are described later, as well as additional mutations in tumor suppressor genes. The complexity of these molecular details can be daunting, but they are important for two clinically relevant reasons:
Prognosis. The mutations found in a particular acute leukemia are often predictive of outcome with conventional therapies.
Targeted therapies. In some instances, the mutated proteins are themselves the therapeutic target.
As targeted therapies become more prevalent, the molecular subtyping of acute leukemia will take on even greater importance. Indeed, it may soon be routine to completely sequence the genomes of acute leukemia cells (as well as other cancers), providing a complete understanding of all of the genetic changes that are responsible for an individual patient’s tumor.
ACUTE LYMPHOBLASTIC LEUKEMIA/LYMPHOBLASTIC LYMPHOMA
Acute lymphoblastic leukemia and lymphoblastic lymphoma encompass different clinical presentations of similar disease entities that are defined by the unbridled proliferation of immature lymphoid cells referred to as lymphoblasts. Acute lymphoblastic leukemia presents in the bone marrow and the peripheral blood, whereas lymphoblastic lymphoma presents as a mass within the bone or soft tissues. These different clinical manifestations relate in part to the origin of the tumor. B-cell tumors almost always arise in the bone marrow (where B cells develop) and present as “leukemias,” whereas T-cell tumors often arise in the thymus (where T cells develop) and present as lymphomatous masses. For simplicity we will discuss these two forms of the disease together under the rubric of acute lymphoblastic leukemia/lymphoblastic lymphoma, or ALL.
ALL is the most common cancer of children. There are about 2500 to 3000 new cases of pediatric ALL in the United States per year. The disease also occurs throughout life in adults but makes up a much smaller fraction of the cancer burden in this segment of the population. The factors leading to the pathogenic mutations that drive ALL development are unknown. It most commonly appears sporadically in previously healthy, normal children. There are two major ALL subdivisions, ALL of B-cell origin (B-ALL) and ALL of T-cell origin (T-ALL). These two types of ALL are morphologically identical but differ in terms of their clinical characteristics, immunophenotype, and genetics.
B-ALL is the most common type of ALL, comprising roughly 85% of cases. The peak incidence is about age 3 years, which closely coincides with the time of peak bone marrow production of B cells during life. Groups that are at relatively increased risk include children of White or Hispanic origin as well as those with Down syndrome (trisomy 21).
The most common clinical features in B-ALL are related to the replacement of the marrow by lymphoblasts and the resulting pancytopenia. The patient usually presents acutely with several weeks of fatigue or listlessness due to increasing anemia, often accompanied by easy bruising due to thrombocytopenia. Neutropenia may lead to infection, producing fever and sometimes localizing signs. Inspection of the peripheral blood film reveals the presence of lymphoblasts, which may be few or numerous, as well as anemia, thrombocytopenia, and granulocytopenia of variable severity. Unusual patients with lymphomatous presentations may complain of pain in a single long bone or come to attention due to involvement of the skin. B-ALL tends to spread via the meninges to the central nervous system and also may involve other immunologically privileged sites such as the ovary and testis. Splenomegaly, hepatomegaly, and lymphadenopathy may also be present due to tumor infiltration, but these features are usually not prominent.
The diagnosis can be strongly suspected based on morphologic inspection of the blood or the marrow. By definition, the diagnosis in those with leukemic presentations requires that lymphoblasts, cells with fine chromatin, small nucleoli, and scant agranular cytoplasm (Fig. 21-2), comprise at least 25% of the marrow cellularity. However, definitive diagnosis requires immunophenotyping, which is usually carried out by flow cytometry. The tumors cells are positive for terminal deoxynucleotidyl transferase (TdT, an enzyme that is expressed only in immature B and T cells) and certain B-lineage proteins such as CD19, and negative for surface immunoglobulin, which appears only on mature B cells (Fig. 21-3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


