Acute Kidney Injury and Chronic Kidney Disease
Robin Y. Beeman and Roberta J. Emerson
Key Questions
• What are the characteristic findings of uremic syndrome?
• How can acute kidney injury be prevented, and how is it treated?
• How is the progression of chronic kidney disease retarded?

http://evolve.elsevier.com/Copstead/
The kidneys have a number of regulatory roles within the body.1 These key functions include the regulation of body fluid volume and osmolality, electrolyte balance, and acid-base balance. Additionally, the kidneys produce and secrete hormones, and excrete metabolic waste products and foreign materials.1 When the kidneys are unable to carry out these functions on a temporary or permanent basis, the ramifications are significant to each body system.
Acute Kidney Injury
Acute kidney injury (AKI), formerly known as acute renal failure (ARF), represents a broad spectrum of kidney diseases ranging from minor changes in renal function to complete renal failure requiring renal replacement therapy.2–5 Acute kidney injury is the sudden reduction of kidney function causing disruptions in fluid, electrolyte, and acid-base balances; retention of nitrogenous waste products; increased serum creatinine level; and decreased glomerular filtration rate (GFR).2–4 Classification criteria for AKI have been developed to identify kidney injury and improve patient outcomes. The 5-point system is known as the RIFLE classification system (R = Risk of injury, I = Injury, F = Failure, L = Loss of function, and E = End-stage kidney disease)2,3 (Table 28-1). The first three stages indicate severity of kidney injury and the last two stages represent patient outcomes. Subsequent to the development of the RIFLE classification, The Acute Kidney Injury Network (AKIN) proposed modifications to the RIFLE criteria, namely, the addition of an absolute increase in serum creatinine concentration of ≥0.3 mg/dl and the specification that the reduction in kidney function occur within a 48-hour period.2
TABLE 28-1
RIFLE CLASSIFICATION FOR STAGING ACUTE KIDNEY INJURY
| STAGE | GFR CRITERIA | URINE OUTPUT CRITERIA |
| Risk | Serum creatinine increased ×1.5 or GFR decreased by 25% | Urine output <0.5 ml/kg/hr for 6 hr |
| Injury | Serum creatinine increased ×2 or GFR decreased by 50% | Urine output <0.5 ml/kg/hr for 12 hr |
| Failure | Serum creatinine increased ×3 or GFR decreased by 75% or serum creatinine >4 mg/dl with acute rise ≥0.5 mg/dl | Urine output <0.3 ml/kg/hr for 24 hr (oliguria) or anuria for 12 hr |
| Loss | Persistent acute kidney failure; complete loss of kidney function >4 wk | |
| End-stage kidney disease | Complete loss of kidney function >3 mo |
From Lewis SL, Dirksen SR, Heitkemper MM, Bucher L, Camera I: Medical-surgical nursing, ed 8, St Louis, 2011, Mosby.
The incidence of AKI in hospitalized patients ranges from 2% to 7%, with higher rates in elderly patients.2,3 Despite improvements in treatments, mortality rates range from 40% to 90%.3 In critical care units, the incidence of AKI reportedly ranges from 1% to 25% with mortality rates between 15% and 60%.6
Etiology and Pathophysiology
The risk of developing acute kidney injury is increased by certain preexisting conditions. These comorbidities include preexisting kidney impairment, cardiovascular and peripheral vascular disease, hypertension, diabetes mellitus, heart failure, malignancies, and benign prostatic hypertrophy.2 Not only are the elderly more likely to have one or more of these conditions, but also aging itself results in changes within the kidney that make it more susceptible to damage. Renal blood flow may drop by 50% between the ages of 20 and 80 and the GFR by about 8 ml/min/1.73 m2 per decade after the age of 30.2,8 These alterations in function increase the risk of AKI and can negatively impact overall prognosis. The aging kidney is less capable of concentrating and diluting urine, conserving sodium, producing prostaglandin, and maintaining renin and aldosterone levels.2,8 See Chapter 26 for a discussion of the effects of aging on renal function.
Acute loss of renal function is attributed to conditions that affect renal perfusion (prerenal), factors that obstruct urine flow distal to the kidney (postrenal), or circumstances within the kidney blood vessels, tubules, glomeruli, or interstitium (intrinsic).2,7 These anatomic delineations are broadly seen as the types or causes of AKI, but the specific etiology must also be identified. Determination of the specific etiology as well as the type of AKI is essential for effective management. Box 28-1 shows the types of AKI and some of their etiologies.
Prerenal Kidney Injury
When acute kidney injury develops because of diminished perfusion of the kidney, it is termed prerenal kidney injury because the etiology occurs before the kidney itself.10 As seen in Box 28-1, this can be due to an absolute or relative decrease in circulating volume, or abnormalities of renal hemodynamics. Actual or relative depletion of volume are the most common etiologies.27 Fever, vomiting, diarrhea, burns, hemorrhage, and overuse of diuretic therapy produce fluid volume deficits that can lead to prerenal kidney injury. Decreased renal perfusion also results if large volumes of fluid collect in extravascular spaces as in edema (interstitial space) or ascites (peritoneal space). Any number of conditions reduces the ability of the heart to generate a cardiac output sufficient to meet the needs of body organ systems. Even though the kidney receives 20% to 25% of the cardiac output,7 that volume may be inadequate when the cardiac output is markedly decreased by cardiogenic shock, heart failure, or lethal ventricular dysrhythmias.
Although most patients who develop prerenal kidney injury have an episode of decreased blood pressure that results in decreased perfusion to the kidney, in some cases perfusion drops without the blood pressure falling below normal.9 These “normotensive” cases of prerenal kidney injury arise in susceptible individuals with very modest reductions in blood pressure who have preexisting impairments in renal autoregulation. Use of nonsteroidal antiinflammatory drugs (NSAIDs), angiotensin-converting enzyme inhibitors (ACEIs), and angiotensin II (AII) receptor blockers is known to interfere with renal vascular autoregulation and can precipitate prerenal kidney injury in certain populations of patients. This includes those who are older than 60 years of age with atherosclerotic cardiovascular disease or have preexisting renal insufficiency (elevated serum creatinine level), heart failure, advanced liver disease, or nephrotic syndrome.2,9 These drugs cause either vasoconstriction of afferent arterioles (NSAIDs) or vasodilation of efferent arterioles (ACE inhibitors and AII blockers); either of these actions results in a decrease in glomerular perfusion pressure.2,9 Thrombus, embolus, dissection, or stenosis of the renal arteries will also result in prerenal kidney injury,2,9 and the risk increases significantly if ACE inhibitors or AII blockers are being used.2,9
Prerenal oliguria is the kidney’s normal physiologic response to a decrease in perfusion,7,9 and at least for a time the renal tissue is unharmed. Neurohumoral mechanisms of local autoregulation are activated as the kidneys attempt to autoregulate perfusion and maintain GFR, and systemic mechanisms such as the renin-angiotensin-aldosterone system (RAAS) act to increase the total circulating volume.2,7 The sensed decrease in renal blood flow results in a decrease in GFR and urine output. Because of the kidney’s ability to tolerate significant reduction in perfusion (as long as it is not greater than 20% to 25% of normal), and as long as the hypoperfusion etiology is identified and corrected, prerenal oliguria will not affect the parenchyma of the kidney.2,7 Efforts to restore adequate perfusion should be fully effective in restoring normal renal function within 1 to 2 days.9 In the case of normotensive patients who have impaired perfusion, by far the smallest subset of prerenal kidney injury cases, interventions must be targeted to the specific etiology.9 Regardless of the etiology, persistent prerenal kidney injury will result in hypoxic renal cells. If hypoxia continues and ischemia lasts more than a few hours, prerenal kidney injury will progress to acute tubular necrosis (intrinsic kidney injury).7,8
Postrenal Kidney Injury
Obstruction of the normal outflow of urine from the kidneys can result in postrenal kidney injury.2,7,8 Box 28-1 lists the most common etiologies of this type of AKI. If only one kidney is affected, the activity of the remaining kidney will increase to maintain fluid and electrolyte balance.2,7 Obstruction of the renal pelvis or ureters of both kidneys, of the bladder outlet, or of the urethra will result in discernible postrenal kidney disease. This type of AKI is the least common and the most amenable to intervention. Normalization of renal function depends on the length of time the obstruction persists.7 Should obstruction persist, the increasing retrograde pressure of urine will result in acute tubular necrosis (intrinsic AKI), and if the obstruction continues over several days or weeks, irreversible damage to the kidney will result.7,8
Intrinsic/Intrarenal Kidney Injury
AKI intrinsic to the kidney itself is further classified by the specific anatomic area involved: vascular, interstitial, glomerular, or tubular2 (see Box 28-1). Some references incorporate the vascular and tubular classifications together because damage to one ultimately leads to damage of the other.7 All of these etiologies are capable of producing the potentially reversible rapid decline in renal function that is AKI. When the small vessels within the kidney are inflamed, obstructed, or damaged by an acute hypertensive episode, the injury may be sufficient to impair nephron functioning. Acute glomerulonephritis is due to an abnormal immune reaction, whereby immune complexes are deposited in the basement membrane of the glomerulus, thereby damaging the glomeruli.7 Normal renal function is disrupted to some degree during this acute inflammatory process, usually lasting about 2 weeks. Acute glomerulonephritis is discussed in detail in Chapter 27. Inflammation of interstitial tissues may be sufficient to result in intrinsic kidney injury. This is usually due to an infection of the kidney (pyelonephritis), an allergic reaction to medications, or an autoimmune disease.2,5 Long-standing pyelonephritis causes damage to the renal medulla and progressive loss of functional renal tissue.7
By far the most common cause of intrinsic kidney injury is acute tubular necrosis (ATN), which itself has many potential etiologies.2,7 Acute tubular necrosis is the result of tubular cell injury, primarily attributable to ischemia or exposure to nephrotoxic substances. Acute tubular necrosis accounts for nearly half of all cases of AKI in hospitalized patients. Sepsis is the most common cause of ischemic ATN and may develop in about 50% of critically ill patients. It causes vasodilation leading to hypoperfusion within the kidney.10 In the elderly, about 30% of ischemic cases are due to sepsis and another third are due to surgical interventions.2 Prolonged prerenal kidney injury, perioperative and postoperative hypotension, hemorrhage, gastrointestinal drainage, and preoperative cardiac complications also contribute to many case of ATN.2
The list of medications and chemicals toxic to the kidney is expansive,2,7 each one inducing a specific toxic reaction in the tubular cells and causing the death of many of them.7 Of all of these nephrotoxins, contrast medium is the most common offending agent.2,7 Contrast-induced AKI (also known as contrast-induced nephropathy) is a major cause of AKI in hospitalized elderly patients, and can develop within 12 to 24 hours of contrast administration.10 Risk factors for developing contrast-induced AKI are underlying kidney insufficiency, age greater than 70, volume depletion, repeated exposures to contrast media in a short time, and coexisting heart failure or diabetes mellitus.10 Prevention is aimed at avoidance of unnecessary contrast administration to high-risk patients, avoidance of multiple procedures over a 24- to 48-hour period, and adequate administration of hypotonic and isotonic intravenous (IV) fluids before and after contrast administration.2,10 ATN caused by contrast media results in prolonged hospitalization, increased health care costs, and an increased risk of death.37 Other nephrotoxic agents include commonly used medications such as aminoglycosides, NSAIDs, amphotericin B, cisplatin, and tetracycline.2,7,10
In ATN, there are two pathophysiologic processes that result in the rapid decrease in glomerular filtration rate: a vascular process and a tubular process.11 The two processes are interrelated, and the severity of one contributes to the severity of the other. Renal blood flow is decreased by 30% to 50% in ATN, and blood is shunted from the medulla to the cortex, further compromising the medullary cells.11,12 Local vasoconstrictors such as prostaglandins and leukotrienes are released, and the effects of sympathetic nervous system (SNS) stimulation contribute further to the vasoconstriction.12 Hypoxia or direct tubular damage, attributable to toxins, initiates an inflammatory response, activating the cascade of inflammatory mediators. When perfusion is restored, more inflammatory cells are enlisted and reperfusion injury perpetuates damage in some areas. Cells in part of the proximal tubule and outer cortex begin the repair process when perfusion is returned, but endothelial cells and those in the ascending limb continue to be injured, become necrosed, and commit apoptosis, resulting in a further decline in GFR.11,12
The pathogenesis of the tubular process is a reflection of the ischemia11 and the inflammatory process (Figure 28-1). Damaged tubular epithelial cells, both viable and nonviable, are shed from the basement membrane and accumulate in the tubular filtrate, where they obstruct filtrate flow.7,11 These cells combine with inflammatory cells and debris to form casts that contribute further to the urinary obstruction.11 Obstructed urinary flow produces an increased pressure within the nephron that is communicated backwards to the glomerulus, further reducing the GFR. The increasing pressure generated by the tubular obstruction forces filtrate through the partially denuded tubular basement membrane into the interstitial space and even into the bloodstream, a process known as tubular backleak.10 Half of the already limited quantity of glomerular filtrate may be lost to the interstitium by this process.
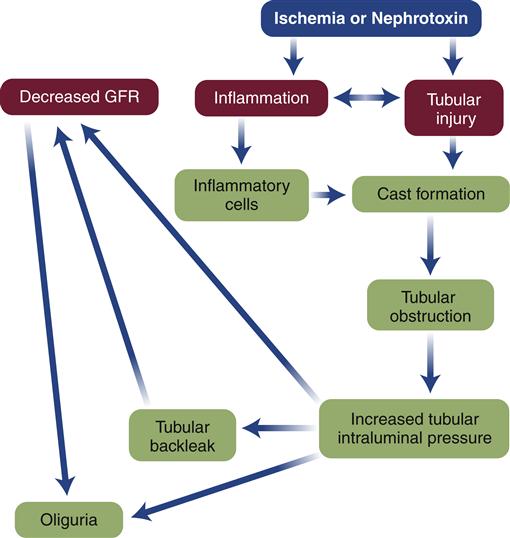
Recovery following ATN is highly dependent on the extent of injury and slower than in the other two types of AKI.7,12 If sufficient destruction of the basement membrane occurs, there may be no recovery and the patient develops end-stage renal disease, the final stage of chronic kidney disease. But the tubules can repair themselves within 10 to 20 days when the basement membrane is intact and new epithelial cells are produced on that surface.7 As with the other types of renal failure, the clinical presentation of ATN is primarily a reflection of the loss of the normal functions performed by the kidney.
Clinical Presentation of Acute Tubular Necrosis
Prerenal kidney injury can be reversed if treated before perfusion drops to below 20% of normal and ischemia occurs.7 It is at this point that ATN develops. Prerenal or postrenal kidney injury will ultimately progress to intrinsic kidney injury if not corrected within a few hours.7 The course of ATN is roughly divided into three phases, and the clinical presentation varies with the phase16 (Figure 28-2). The laboratory findings that differentiate prerenal from intrinsic kidney injury are shown in Table 28-2. Table 28-3 provides the laboratory profile associated with renal failure; some of these findings are more likely to be noted in end-stage renal disease than in AKI. Though serum creatinine levels begin to increase within 12 hours to 2 days after injury,3 new biomarkers are being investigated to detect AKI earlier than the rise in serum creatinine level, with the promise of leading to earlier detection and treatment. Examples of these new biomarkers currently being tested are interleukin-18, neutrophil gelatinase-associated lipocalin, and kidney injury molecule-1.3,17As with other aspects of the clinical presentation, laboratory findings are affected by the phase of ATN. Although the naming of the phases of AKI varies among resources, the clinical progression of ATN is consistent.
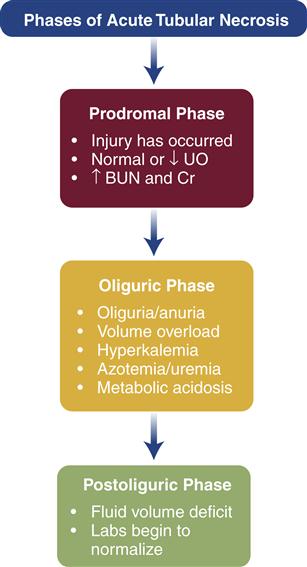
TABLE 28-2
LABORATORY VALUE DIFFERENCES IN PRERENAL AND INTRINSIC AKI
| LABORATORY TEST | PRERENAL FINDINGS | INTRARENAL FINDINGS |
| FENa % | <1 | >2 |
| Proteinuria | Absent | Possible |
| Urine specific gravity | >1.020 | 1.010-1.020 |
| Urine osmolality (mOsm/kg) | >500 | 300-500 |
| BUN/creatinine ratio | >20:1 | 10-20:1 |
| Urine sodium concentration (mmol/L) | <10 | >20 |
| Urinary sediment | Few hyaline casts | Tubular, RBC, and WBC casts |
∗Derived from Gammill HS, Jeyabalan A: Acute renal failure in pregnancy, Crit Care Med 33(10):S372-S384, 2005; Needham E: Management of acute renal failure, Am Fam Physician 72(9):1739-1746, 2005; Lameire N, Van Biesen W, Vanholder R: Acute renal failure, Lancet 365:417-430, 2005.
AKI, Acute kidney injury; FENa %, fraction of excreted sodium, percent.
TABLE 28-3
LABORATORY PROFILE FOR RENAL DISEASE
| TEST | NORMAL RANGE FOR ADULTS | VALUES IN RENAL DISEASE | COMMENTS |
| Test to Evaluate Removal of Nitrogenous Wastes | |||
| Serum creatinine | |||
| Blood urea nitrogen | |||
| Electrolyte Studies | |||
| Serum sodium | 136-145 mEq/L; 136-145 mmol/L (SI units) | Normal or decreased | |
| Serum potassium | 3.5-5.0 mmol/L (SI units) | Increased | |
| Serum phosphorus (phosphate) | Increased | ||
| Serum calcium | Decreased | ||
| Serum magnesium | 1.3-2.1 mEq/L; 0.65-1.05 mmol/L (SI units) | Increased | Advise patient to avoid compounds containing magnesium (e.g., laxatives). |
| Serum bicarbonate | 23-30 mEq/L (venous); 23-30 mmol/L (SI units) | Decreased | |
| Arterial blood pH | 7.35-7.45 | Decreased (in metabolic acidosis) or normal | |
| Arterial blood bicarbonate (HCO3−) | 21-38 mEq/L | Decreased | Provide replacement oral, IV, or by hemodialysis or peritoneal dialysis. |
| Arterial blood (PaCO)2 | 35-45 mm Hg | Decreased | Monitor for respiratory fatigue (client breathes more rapidly and deeply to “blow off” carbon dioxide). |
| Other Blood Studies | |||
| Hemoglobin | Decreased | ||
| Hematocrit | Decreased to 20% | ||
| Urinalysis∗ | |||
| Specific gravity | Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||