Acute Disorders of Brain Function
Joni D. Marsh and Jacquelyn L. Banasik
Key Questions
• What are the proposed mechanisms and potential consequences of secondary brain injury?
• What are the common long-term sequelae of stroke, and how are they managed?
• What are the causes and usual presentations of cerebral aneurysm and arteriovenous malformation?

http://evolve.elsevier.com/Copstead/
Disorders of brain function can result from a wide variety of pathophysiologic processes. The focus of this chapter is on primary causes of acute brain injury including brain trauma, cerebrovascular disease, brain hemorrhage, and central nervous system (CNS) infections. These conditions are acute because they generally have a sudden onset and progress rapidly. Thus, early detection and prompt management are necessary to prevent death and minimize morbidity. However, the majority of patients who survive acute injury to brain structures will be left with some degree of permanent neurologic damage and chronic dysfunction. The designation of acute and chronic disorders is, therefore, somewhat arbitrary. The chronic aspects of neurologic diseases, including seizure disorders and dementias, are discussed in Chapter 45. Acute neurologic dysfunction often is a complication of diseases primarily affecting other systems. Hypoglycemia, renal failure, liver failure, human immunodeficiency virus infection, drug overdoses, fluid imbalances, and many other abnormalities may cause acute brain dysfunction. Accurate determination of the source of acute alterations in brain function is an important step in developing an appropriate treatment plan. However, there are many common elements in the pathogenesis of acute brain injuries, regardless of etiologic factors. These cellular aspects are presented as a foundation for understanding the specific disorders that follow.
Mechanisms of Brain Injury
The mechanisms of brain injury are varied, complex, and incompletely understood. Mechanical trauma, ischemia, cellular energy failure, reperfusion injury, excitotoxins, edema, vascular failure, and injury-induced apoptosis (programmed cell death) are all factors thought to be operative in most kinds of acute brain injury. These mechanisms are often separated into two categories: primary injury and secondary injury.
The primary injury is that which occurs immediately at the onset of brain injury. This definition implies that there is little that can be done to reverse the process once it has occurred. For example, in the case of head trauma, some tissues will be irreversibly damaged at the time of impact owing to mechanical forces. This damage represents the primary injury. Similarly, with the sudden cessation of blood flow to an area of brain tissue, as occurs in stroke, an area of irreversible ischemia in cells may develop quickly, and this constitutes the primary injury. Brain tissue necrosis occurs rapidly as cells lose membrane integrity, rupture, and release their intracellular contents into the extracellular space. Cytotoxic edema quickly follows, which can cause deleterious effects to surrounding brain tissue.
Secondary injury refers to the development of further neurologic damage subsequent to the primary injury, and this may progress over days or weeks. Delayed cell death may involve necrosis from further acute injury or may be a delayed consequence of the primary injury. Cells that die slowly after injury are said to undergo apoptosis or programmed cell death. Apoptosis requires energy and protein synthesis, and the cells shrink and die in a tidy fashion without releasing their internal contents. Necrosis follows severe ischemic injury, whereas apoptosis is associated with moderate injury. A critical factor in determining the neuronal cell fate after injury is the degree of adenosine triphosphate (ATP) depletion. If ATP levels fall profoundly, increased membrane permeability and necrosis ensue. If the ATP level is partially maintained for a period of time after the injury, apoptosis is the likely consequence. Mild reductions in the amount of ATP are associated with reversible injury and cellular recovery.1 ATP level reduction is a consequence of ischemia and hypoxia, which accompany many types of acute brain injury including trauma and stroke.
Mechanisms of secondary injury are the subject of much interest because of the potential to effectively intervene to limit brain damage. Unfortunately, effective means of preventing secondary damage have remained elusive, leading to high rates of mortality and morbidity. The effort to elucidate mechanisms of secondary injury and develop effective treatments would seem worthwhile because the degree of primary injury is thought to be small in most cases.2 Thus, the high rates of mortality and morbidity may be attributed in large part to mechanisms of secondary injury.
Ischemia and Hypoxia
Ischemia occurs when the delivery of oxygenated blood is below the level needed to meet metabolic demands of the brain tissue. Ischemia is a contributing factor in most forms of acute brain injury, either as the primary insult (e.g., stroke) or as part of the secondary response to injury (e.g., vasospasm, vascular compression, or abnormal autoregulation). Hypoxia is a deficiency of oxygen at the cellular level, which may occur as a result of decreased blood flow (ischemia) or decreased blood oxygenation (hypoxemia). In practice, ischemia and hypoxia usually occur together and are considered together in this discussion. Ischemia results in immediate neurologic dysfunction because of the inability of neurons to generate the ATP needed for energy-requiring processes. In addition, ischemia sets the stage for secondary injury by oxygen free radicals, excitatory amino acids, and inflammatory cells.
Cellular Energy Failure
Neuronal tissue is highly sensitive to oxygen deprivation because it has great ATP requirements and limited capacity for anaerobic metabolism during ischemia. The normal brain receives about 15% of the total cardiac output and garners 20% of the body’s oxygen consumption (750 ml/min), despite contributing only 2% of the body weight.1 Neurons are dependent on glucose for production of ATP; however, they store little in the form of glycogen. Thus, when oxygen supply is decreased, not only is oxidative phosphorylation impaired, but also the low supply of stored glucose restricts anaerobic production of ATP. Brain cells tolerate loss of ATP for several minutes; about 5 to 10 minutes of complete occlusion is necessary for irreversible brain damage in humans. Complete occlusion of blood flow is rare but even a partial occlusion, if allowed to continue for a sufficient amount of time, may produce irreversible brain damage. Once blood flow to cerebral neurons diminishes, two mechanisms can independently lead to brain cell death: anaerobic metabolism and deterioration of ion gradients.
A general sequence of events following acute brain ischemia has been proposed (Figure 44-1).3 The critical event is mitochondrial dysfunction owing to lack of cellular oxygen. Recall that oxygen is required to accept electrons from the mitochondrial electron transport chain. In the absence of oxygen, the transport proteins and cytochromes remain reduced and unable to accept any more electrons from the Krebs cycle (tricarboxylic acid cycle). Anaerobic glycolytic pathways are initiated in the affected region to compensate for the loss of oxygen and to provide a source of energy. Glycolysis may continue for a short time, producing pyruvate, which is converted to lactate. However, this conversion releases H+ and contributes to cellular acidosis, a damaging by-product of glycolysis. Toxicity of hydrogen ions leads to loss of neuronal integrity.
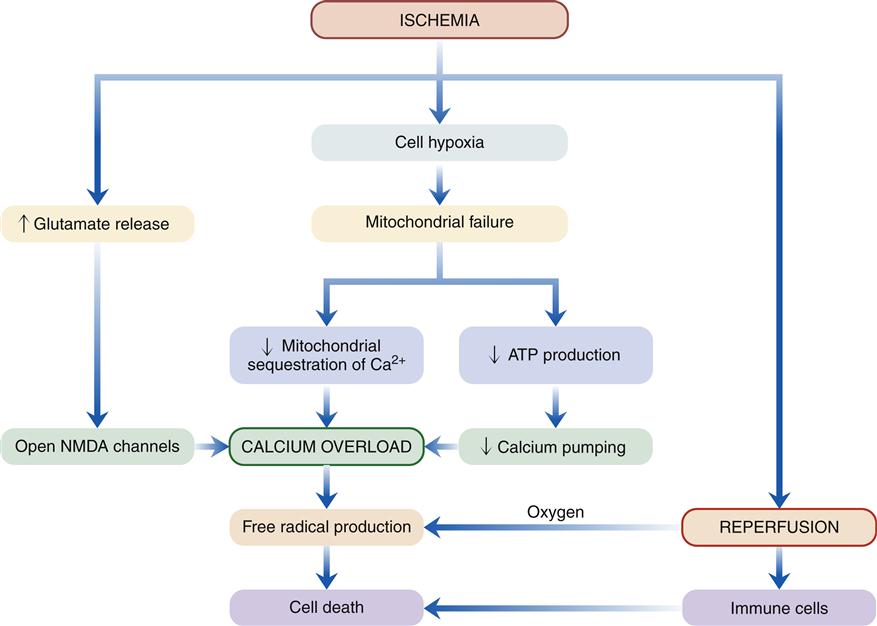
Inadequate energy supply leads to deterioration of ion gradients. Most of the ATP used by neurons is for maintenance of ion gradients across the plasma membrane. The sodium-potassium (Na+-K+) pump consumes three fourths of the ATP in a typical neuron. Anoxic depolarization causes potassium to leave the cell and sodium, chloride, and calcium ions to enter. Energy also is required to maintain calcium balance and regulate neurotransmitter synthesis and reuptake. Not surprisingly, energy failure results in neuronal dysfunction, injury, and, if severe or prolonged, necrotic cell death. Ischemia also is the probable inciting factor for apoptosis (see Chapter 4).
The mitochondria are also important regulators of calcium ion concentration in the cell. The mitochondrial membrane contains calcium transporters that sequester calcium ions within the mitochondria when cytoplasmic calcium levels are elevated. Mitochondrial energy failure impairs the ability of mitochondria to perform this sequestering function. Thus, the mitochondria become severely overloaded with calcium, which activates enzymes (phospholipases) that damage mitochondrial membrane structures. Ischemic cells are prone to calcium overload because pumps that move calcium out of the cell are energy dependent. Calcium ions have a large electrochemical gradient for diffusion into the neuron and tend to accumulate intracellularly.
Unfortunately, the activity of many intracellular enzymes is regulated by intracellular calcium. Calcium overload is thought to be a critical factor leading to activation of enzyme cascades, which disrupt function and cause irreversible damage to cell membranes (lipid peroxidation). One might speculate that measures to inhibit calcium entry into damaged cells would be of therapeutic benefit. One way to reduce calcium influx is by administration of calcium channel–blocking agents. These drugs block voltage-gated calcium channels. Unfortunately, clinical trials with calcium channel–blocking agents have failed to show benefit.2 Other avenues for inhibiting calcium overload are being investigated, including those that block the effect of glutamate receptors as described in the following section.
Excitatory Amino Acids
Calcium may gain entry into cells by portals other than voltage-gated channels. Glutamate is an excitatory amino acid neurotransmitter thought to be important in learning and memory. Overstimulation of neurons by glutamate is associated with cell injury, leading to its designation as an excitotoxin. Glutamate binds two kinds of receptors that are linked to the opening of ion channels in the plasma membrane of neurons. N-methyl-D-aspartate (NMDA) receptors have received the most attention, but α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) channels are also thought to contribute to the neurotoxic effects of glutamate. Activation of AMPA receptors results in opening of Na+ channels in the membrane, which leads to depolarization. Depolarization then affects the NMDA channels by removing a Mg2+ ion that usually blocks the NMDA channel (Figure 44-2). Subsequent binding of glutamate to the NMDA receptor opens it and allows Ca2+ to enter the cell accompanied by inflow of water, which results in cytotoxic edema, a rapid swelling of neurons. As previously described, calcium overload mediates a cascade of events leading to cell injury.
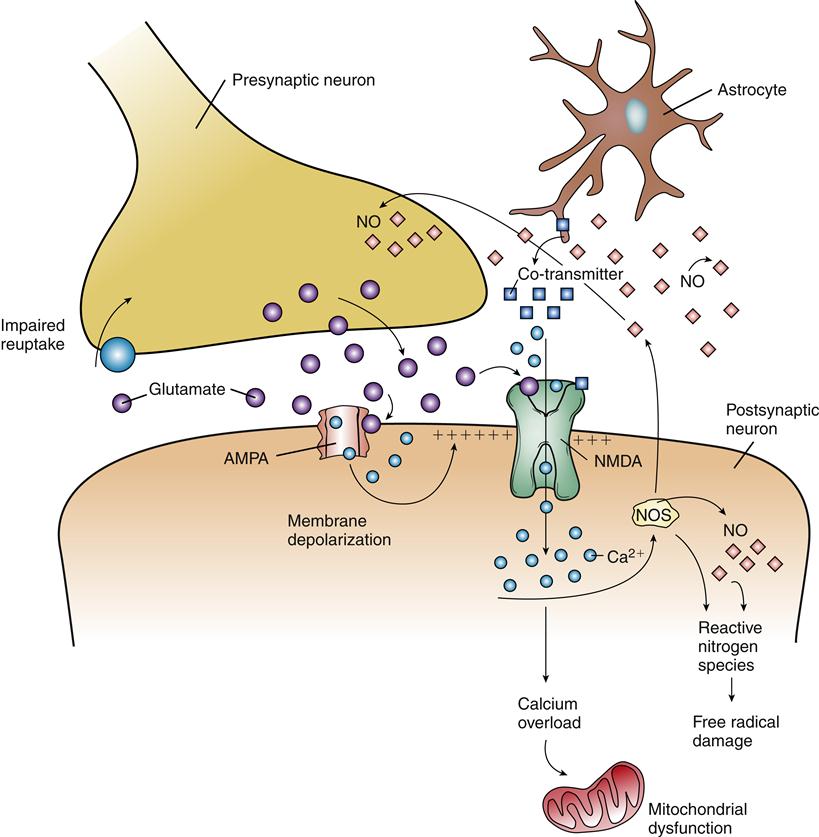
The amount of glutamate in the synapses is usually tightly regulated by release and reuptake controls. In the presence of neuronal injury, excessive glutamate may be released because of impaired membrane integrity. With concomitant ischemia, reuptake mechanisms fail to remove excess glutamate from the synapse because they are energy-dependent processes. Excess glutamate stimulates nearby neurons, which then take up large amounts of injurious calcium ions. Small neurons in the cerebral cortex and hippocampus are particularly prone to glutamate excitotoxicity, and selective damage in these areas may occur. In addition to the calcium overload mechanism of injury, NMDA receptor activation stimulates nitric oxide (NO) production in neurons. NO is a neurotransmitter, but in excess it may increase the production of reactive nitrogen species (RNS), which function as free radicals to damage cellular components. The potential neuroprotective effects of controlling glutamate release or activity have been investigated but have not clearly shown efficacy in improving outcomes.2
Reperfusion Injury
Reestablishing perfusion to an area of prior ischemia is a matter of great urgency if neuronal tissue is to be salvaged. The longer and more severe the period of ischemia, the greater the extent of necrosis. However, previously ischemic cells face new dangers with the return of blood flow. In particular, the return of oxygen brings the potential for oxygen free radical formation, and the flow of blood allows inflammatory cells to invade the area. The secondary injury that occurs after reestablishing blood flow has been termed reperfusion injury and has been studied extensively in cardiac tissues. The mechanisms in the brain appear to be similar. During the period of ischemia, brain cells accumulate substrates for oxidative phosphorylation, including the free radical–forming metabolites of adenosine monophosphate (AMP), xanthine, and hypoxanthine. When oxygen reenters the cell, erratic transfer of electrons to oxygen can produce a number of reactive oxygen products that behave as free radicals, damaging cell structures. These include hydroxyl radicals (OH•), superoxide (O2−), and peroxide (H2O2). Cell membranes may undergo lipid peroxidation in response to free radical damage, with subsequent formation of arachidonic acid. The arachidonic acid cascade yields more oxygen free radicals as well as mediators of inflammation.2
The role of immune mechanisms in reperfusion injury of brain tissue is only partially understood. Previously, the CNS was thought to be relatively shielded from immune cells because of the low permeability of the blood-brain barrier (BBB). However, the BBB is believed to be compromised with ischemia because the capillary endothelial cells are injured. The amounts of inflammatory cytokines, including interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor (TNF), increase in the brain during ischemic injury and are thought to attract neutrophils to the area and contribute to brain inflammation.4 The importance of neutrophil recruitment to secondary injury remains controversial, but it may contribute further free radical generation and vascular obstruction by aggregated neutrophils. Trauma and inflammation also promote platelet aggregation in cerebral vessels with subsequent reduction in perfusion and worsening of ischemia.
Abnormal Autoregulation
Under normal conditions, blood flow through brain tissue is controlled primarily by autoregulation. Cerebral vessels respond to metabolic factors including pH, carbon dioxide concentration, and oxygen levels. Cerebral blood flow is closely matched to metabolic needs despite wide fluctuations in perfusion pressure. Blood flow is maintained at a fairly constant rate over a range of mean arterial pressure (MAP) from about 50 to 150 mm Hg.1 Above and below these levels, autoregulation fails. Hypotension predisposes to ischemia, whereas severe hypertension may lead to vascular damage and brain edema.
Appropriate autoregulation is necessary to provide a steady supply of oxygen and nutrients to brain cells and to remove metabolic wastes. Cerebral vessels dilate when mean arterial blood pressure falls or when brain metabolism increases. Anything that interferes with the ability of the vessels to dilate can lead to ischemia. Thrombi, emboli, vasospasm, neutrophil aggregation, and tissue edema may inhibit vasodilating autoregulatory mechanisms. Alternatively, vascular injury may impair vasoconstricting mechanisms and allow hyperperfusion and edema formation. Depending on the cause, autoregulation may be impaired locally, as in an area of thrombosis, or globally, as in generalized cerebral edema.
Autoregulation is influenced by the partial pressures of carbon dioxide (Paco2) and oxygen (Pao2) in the arterial blood. The response to a change in Paco2 is very brisk: as Paco2 levels fall, cerebral vessels constrict, and as Paco2 levels rise, the cerebral vessels dilate. A rise in Paco2 can increase cerebral blood flow significantly. The response to changes in Pao2 is much less dramatic.
The autoregulatory response to Paco2 remains robust, except in severely brain-injured patients, and can cause detrimental increases in cerebral blood flow when respiratory compromise leads to hypercapnia. Excessive cerebral blood volume can exacerbate cerebral edema. Conversely, hyperventilation to produce low Paco2 results in prompt vasoconstriction and, often, a reduction in intracranial pressure (ICP). Hyperventilation had been used for many years as standard therapy in the treatment of patients with increased ICP. However, prolonged hyperventilation does more harm than good because it critically reduces cerebral blood flow to responsive vessels and triggers tissue ischemia in these areas.
Loss of matching between oxygen supply and demand occurs when autoregulatory mechanisms fail. Cerebral oxygen demand is correlated with the degree of neuronal activity and may vary widely in different regions within the brain. Excessive levels of catecholamines or excitatory amino acids can significantly increase cerebral metabolism. In the context of impaired blood flow, these neurotransmitters may contribute to ischemia by increasing cerebral oxygen demand. Likewise, seizure activity increases neuronal metabolism and leads to worsening neurologic outcomes.5
Efforts to reduce release of excitatory neurotransmitters through hypothermia, rest, and pain control may be beneficial. Pharmacologic suppression of brain seizures is imperative in the patient with cerebral ischemia. Drug-induced coma, with agents such as barbiturates, has been advocated to reduce brain metabolism. However, this treatment is not without side effects.2
Hypothermia is a strategy for reducing brain metabolism and protecting the brain from ischemic injury. A number of animal and human studies suggest that cerebral injury can be delayed, and possibly avoided, by cooling the brain.6 Possible mechanisms include inhibition of glutamate release, inhibition of IL-1 release, reduced cerebral metabolic rate, and suppression of inflammation. The effects of hypothermia on patient outcomes and the optimal degree of hypothermia remain controversial. Moderate degrees of cooling (28° to 32° C) are associated with platelet dysfunction and coagulopathy.6 Shivering negates the usefulness of hypothermia by increasing oxygen demand, and must be suppressed, usually by pharmacologic means. Further research is ongoing to determine the therapeutic window for effectiveness, appropriate duration, and safe rewarming protocols.6,7
Two related concepts important to the discussion of autoregulation are cerebral edema and increased ICP. Swelling and space-occupying lesions (mass lesions), such as tumors or hematomas, may increase the pressure within the cranium such that blood supply is compromised. Measures to reduce cerebral edema, remove mass lesions, and prevent elevations of ICP help to maintain functional autoregulation.
Increased Intracranial Pressure
ICP is the pressure exerted by the contents of the cranium, and it normally ranges from 0 to 15 mm Hg.1 Elevated ICP may occur in most types of acute brain injury and is associated with impaired neurologic function attributable to compression of brain structures. In all but very young children, the skull is a rigid, closed system with a set volume and a finite ability to accommodate changes in volume before elevations in ICP occur.
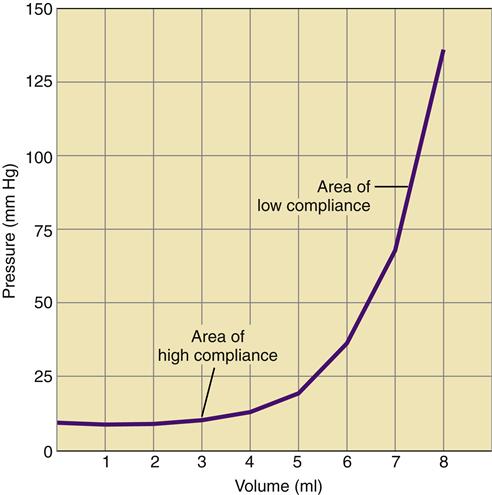
The volume of the cranium is made up of three components: brain tissue, cerebrospinal fluid (CSF), and blood. A relationship known as the Monro-Kellie hypothesis describes the compensatory responses to a change in volume in any of the three components.8 A slight increase in one component can be offset by a reduction in the volume of the other two. An increase in brain volume, as might occur with cerebral edema, can be offset by a reduction in the CSF space and the space occupied by the cerebral vasculature. The ability to accommodate changes in volume without significant increases in pressure is called compliance. Intracranial compliance is limited because of the rigid skull; although small increases in intracranial volume may be absorbed, moderate changes result in significant increases in ICP (Figure 44-3).
Each of the cranial components has a varying capability to compensate for the others. Cerebral blood vessels can reduce their volume through vasoconstriction. The CSF compartment is capable of significant reduction in volume by shunting CSF to the spinal cord or into the venous system via the arachnoid villi. The brain parenchyma has little ability to reduce its volume to compensate for CSF or blood volume expansion. In young children, an increase in ICP may manifest as an increase in head circumference. This occurs because the cranial bones have not yet fused, and the skull can expand to accommodate the increased intracranial volume.
In the healthy brain, transient changes in ICP are common and well tolerated. Sneezing, coughing, straining, and head-dependent positions all cause elevated ICP, but they are without consequence because ICP quickly returns to normal. In the brain-injured person, however, transient elevations in ICP can be very dangerous and poorly tolerated.
Etiology
The most common causes of increased ICP include stroke, trauma, and tumors, but many other primary and secondary disorders can cause significant elevations in ICP (Box 44-1). These disorders have common features in that they all affect the volume of CSF, blood, or brain tissue. An increase in brain tissue volume commonly occurs from conditions that cause cerebral edema. Edema of brain tissues is due to accumulation of fluid in interstitial or intracellular spaces.
Interstitial edema is usually secondary to an increase in capillary pressure, damage to the capillary endothelium from a chemical injury, or a sudden increase in vascular pressure beyond autoregulatory limits. This type of edema has been termed vasogenic, and it results in extravasation of electrolytes, proteins, and fluid into the intercellular space. Vasogenic edema is a consequence of stroke, ischemia, and severe hypertension, and may occur surrounding brain tumors. The edema is often localized to a particular brain region where the BBB has been disrupted. Thus the swelling may be unilateral, occurring in one brain hemisphere or the other. Unilateral swelling often is poorly tolerated because midbrain structures are compressed, and can lead to shifting of cerebral tissue or brain herniation.
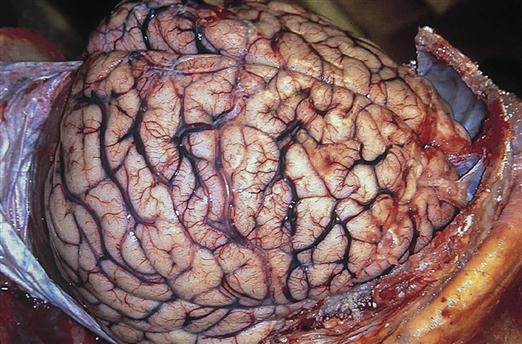
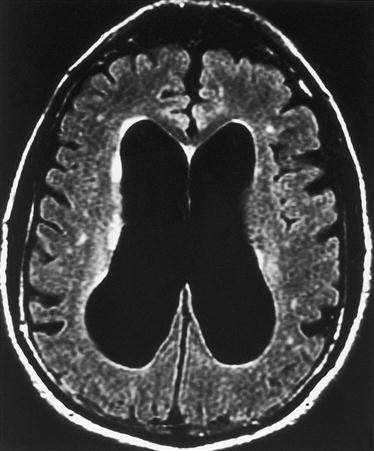
Intracellular edema, called cytotoxic edema, occurs when ischemic tissue swells because of cellular energy failure. A lack of ATP allows Na+ to accumulate in the cell, creating an osmotic force to draw in water. Cytotoxic edema predominates in cases of global ischemia. Global ischemia occurs when oxygenation of the whole brain is impaired, as would occur with cardiac arrest or severe hypoxemia. Generalized brain edema flattens the gyri and reduces the spaces between them (Figure 44-4).
In many cases of acute brain injury, vasogenic and cytotoxic edema occur together. Cerebral edema, when severe, can start a cyclic process that promotes further edema of increasing severity and contributes to increased ICP. As edema fluid collects, it compresses local vessels, preventing adequate blood and oxygen from reaching the cells. This results in ischemia, which in turn triggers vasodilation and increased capillary pressure, further fluid leakage into the injured tissue, and increased edema. Vasogenic edema tends to be a delayed process in terms of the secondary effects of brain injury, progressively worsening during the first several days after injury. Clearance of brain edema occurs primarily by absorption into the CSF system.1
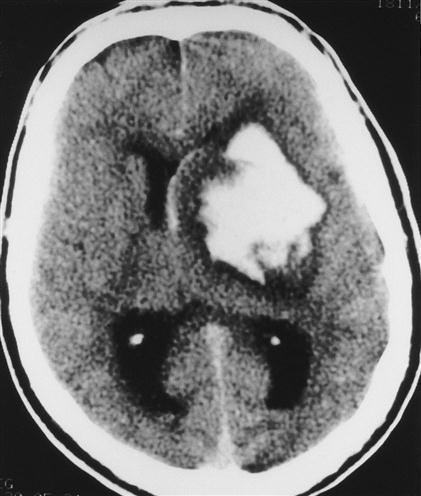
In addition to edema, a number of space-occupying processes, such as tumors, hematomas, and abscesses, can increase intracranial volume and contribute to elevated ICP. These mass lesions are often unilateral and may result in severe compression of vital brain structures. Attempts by the brain to accommodate the expanding mass result in typical findings on computed tomography (CT) scans (Figure 44-5). The ventricles are reduced in size, and midline structures are displaced.
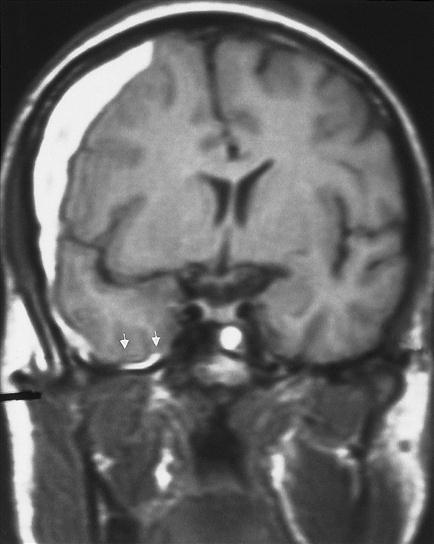
Excessive accumulation of CSF (hydrocephalus) is another important cause of increased ICP. Elevated CSF volume causes the ventricles to enlarge and press on cerebral brain structures (Figure 44-6). Hydrocephalus may be a primary disorder or may develop as a result of obstruction proximal to the arachnoid granulations (obstructive; noncommunicating hydrocephalus) or at the level of the arachnoid granulations (communicating; nonobstructive hydrocephalus). Obstructive hydrocephalus commonly occurs when a lesion blocks the flow of CSF out of the ventricle, whereas nonobstructive hydrocephalus is common following subarachnoid hemorrhage because residual blood clogs the arachnoid villi and prevents the CSF from being reabsorbed.
Increased intravascular cerebral blood volume is unlikely to be a primary cause of high ICP, but it may contribute to pressure elevations initiated by ischemia or trauma. High Paco2 or loss of autoregulatory controls can lead to vasodilation and increased cerebral blood volume.
Manifestations
Manifestations of elevated ICP include headache, vomiting, and altered level of consciousness (drowsiness). The patient may complain of blurry vision, and evaluation of the fundi may reveal edema of the optic disk (papilledema). As ICP rises to higher levels, the level of consciousness decreases, and pupil responsiveness to light becomes impaired. Eventually the patient will exhibit altered respiratory patterns and will become unresponsive to stimulation and unable to move, verbalize, or open the eyes. Prolonged elevations of ICP are thought to damage brain structures by compressing the blood supply and causing ischemia.
Patients exhibiting manifestations of elevated ICP, or those with significant risk for elevated ICP, may be monitored with a pressure device inserted into the brain parenchyma through an opening in the skull (burr hole). The pressure device is connected to an electrical transducer, and the ICP waveforms can be monitored continuously (Figure 44-7). In general, a high ICP is associated with poor outcome. Different ICP waveforms are thought to carry different prognoses. The normal ICP waveform is characterized by three pressure peaks called P1, P2, and P3. These waves are reflections of changes in ICP associated with each arterial pulsation. Normally P1 is higher than P2, and P2 is higher than P3 (see Figure 44-7). As ICP begins to increase, the pattern of waves remains normal, but the peak and mean pressures are higher. Further increases in ICP are characterized by a P2 wave that exceeds P1 and a dampening of the individual waveforms (plateau or rounded waves).9
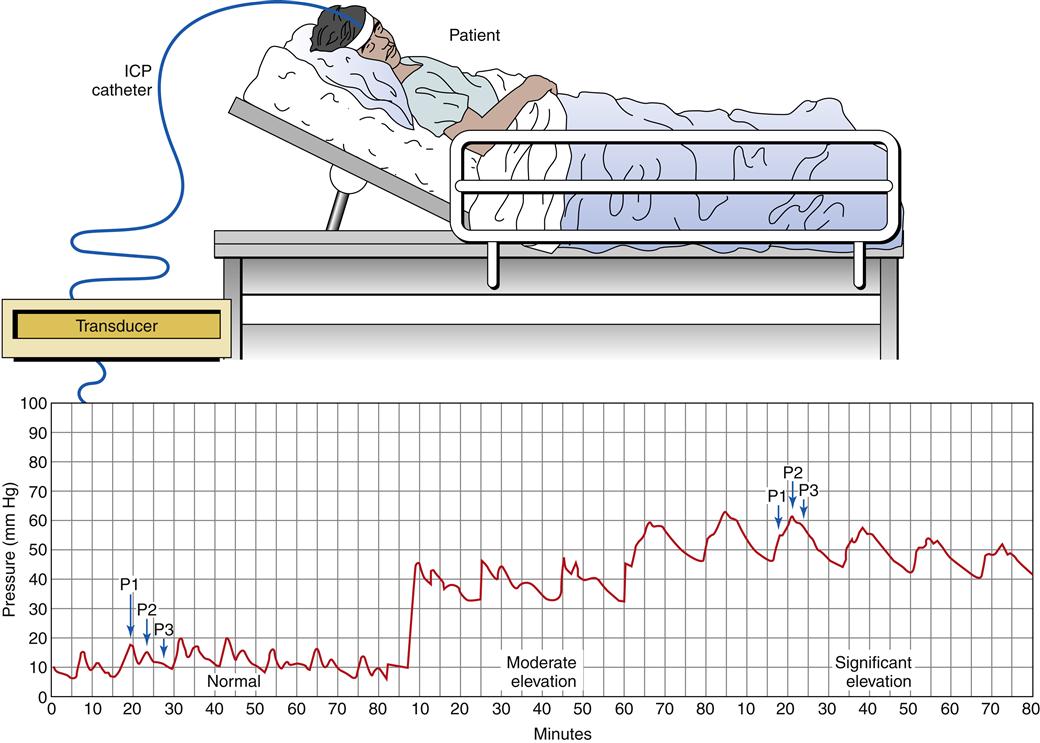
Rounded or monotonous waves reflect severe pathologic increases in ICP attributable to changes in cerebral volume. Plateau waves can reach 50 to 100 mm Hg. After the plateau period the ICP slowly decreases, but it usually remains elevated above baseline. These waves reflect a potentially life-threatening situation, and if the pathologic process is not stopped, a cycle of increased ICP followed by vasodilation to maintain constant blood flow through swollen tissues continues, which in turn further increases ICP. An extreme increase in ICP can precipitate an intense reaction by the sympathetic nervous system as it attempts to maintain cerebral perfusion through the compressed blood vessels. This has been termed an ischemic response or Cushing reflex. The systolic blood pressure can jump to values exceeding 200 mm Hg, accompanied by bradycardia and a widening pulse pressure. The Cushing reflex generally is viewed as a “last ditch” effort by the brain to reestablish cerebral perfusion.8
Brain Compression and Herniation
A dreaded complication of elevated ICP is brain compression and herniation. Compression of midbrain and brainstem structures is associated with rapid neurologic demise unless corrected quickly. Important midline structures include the reticular activating system (RAS), which is necessary for maintaining consciousness, and vital regulatory centers for cardiovascular and respiratory control. Radiologic examination by computerized tomography (CT) scan or other means (e.g., magnetic resonance imaging [MRI]) is useful in evaluating the patient with increased ICP who exhibits a change in neurologic status. CT scans may show midline shifts and herniations when ICP is sufficiently elevated.
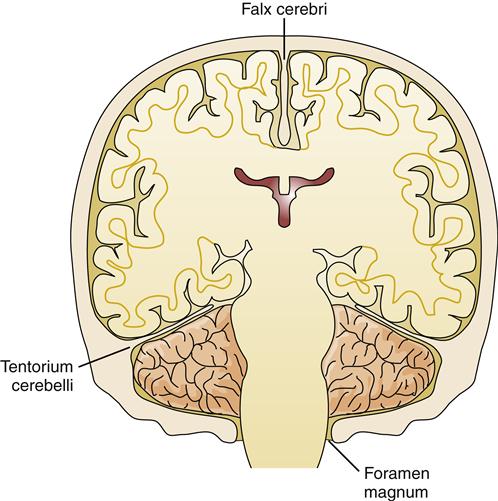
Herniation refers to the protrusion of brain tissue through an opening in the supporting dura of the brain. Several types of brain herniation have been described according to their anatomic locations. The brain parenchyma is divided into compartments by the supporting structure of the dura. The dura folds into the space between the cerebral hemispheres to form the falx cerebri and folds in from the lateral aspects to form the tentorium, which separates the cerebellum from the cerebral hemispheres (Figure 44-8). The most common herniations occur through openings in these structures (Figure 44-9).
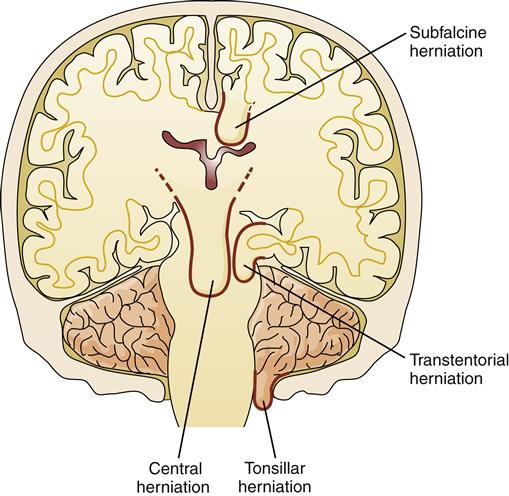
There is a small space between the tip of the falx cerebri and the corpus callosum through which the neurologic lobe of the cerebral cortex can herniate (Figure 44-10). This is called a subfalcine hernia. Subfalcine herniation occurs when a lesion in one hemisphere is large enough to cause a lateral shift across the midline of the intracranial cavity, forcing the neurologic gyrus under the falx cerebri. This results in distortion and compression of the internal cerebral vein. Subfalcine herniation can be asymptomatic and generally carries a better prognosis than other types of brain herniation. The greatest danger results from compression of blood vessels, particularly the ipsilateral anterior cerebral artery, which can cause further cerebral ischemia and edema and contribute to the ICP elevation.
The tentorium is a rigid dural fold that separates the cerebellum and cerebral hemispheres. Midbrain structures pass between the infoldings of the dura in a structure called the incisura. With transtentorial herniations, a part of the brain protrudes through this space. Tentorial herniations are of two types: (1) bilateral herniations, which cause central transtentorial herniation, and (2) lateral herniations, in which one hemisphere compresses midbrain structures to the side and herniates through the tentorial opening (see Figure 44-9).
Central tentorial herniation results from expanding lesions in the frontal, parietal, and occipital lobes that force a downward displacement of the hemispheres and basal nuclei with compression of the diencephalon and adjoining midbrain. Transtentorial herniation can occur rapidly or slowly, depending on the type of lesion. The speed with which the process is recognized is a critical factor in patient survival. Slowly dilating or odd-shaped pupils is an ominous sign that indicates compression of the third cranial nerve and midbrain. Transtentorial herniations are associated with significant intracranial hypertension and may initiate vascular compression and CSF obstruction, which then contribute to the existing problem of ischemia and hypertension.
Uncal herniation is a type of tentorial herniation that typically occurs with expanding lesions in the temporal lobe. As the lobe shifts, the basal edge of the uncus and the hippocampal gyrus bulge over the edge of the incisura (see Figure 44-9). In the process, the third cranial nerve and the posterior cerebral artery are compressed. The pupil on the same side (ipsilateral) as the lesion often becomes dilated and unresponsive to light (fixed). Flattening of the midbrain interferes with the ascending RAS and depresses the level of consciousness. The ipsilateral cerebral peduncle is also compressed, resulting in contralateral motor dysfunction. Compression of the contralateral cerebral peduncle is also common, leading to the confusing symptom of ipsilateral motor dysfunction.
Tonsillar herniation is less common than the other herniation syndromes and involves the shift of the cerebellar tonsils through the foramen magnum and compression of the medulla and upper cervical cord (see Figure 44-9). This typically occurs in patients with cerebellar lesions. Because of the proximity of the cerebellum to the brainstem, tonsillar herniation evolves very rapidly and can result in death in a matter of minutes. Signs usually include precipitous changes in blood pressure and heart rate, small pupils, disturbances in conjugate gaze, ataxic breathing, and quadriparesis.
Management
Management of increased ICP is often based on the results of CT or MRI. Processes amenable to surgical intervention can be detected and treated. Removal of excess CSF, tumors, abscesses, and hematomas can dramatically improve ICP. Nonsurgical processes such as cerebral edema, intracerebral bleeding, and infections are managed medically. ICP measurements and determinations of cerebral perfusion pressure (CPP) are used to guide therapy. Controversy regarding the appropriate treatment of increased ICP continues, and the focus of management has shifted from ICP control to management of cerebral oxygenation. The roles of previously established therapies such as hyperventilation, brain dehydration with diuretics, head-up and neutral body positions, and corticosteroid administration have been called into question. Previously discarded therapies, such as hypothermia, hypertonic saline infusion, and drug-induced comas, have been reintroduced. Despite these controversies in medical management, the value of careful observation and assessment of neurologic function is unquestioned. Many times subtle changes in neurologic function are detectable early in the process of evolving brain injury. Several tools have been developed to help standardize neurologic examination and are discussed in the following section.



