INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Explain the mechanisms underlying antibody-mediated autoimmune hemolysis.
Understand the causes of immune hemolytic anemia as well as its diagnosis and treatment.
Describe the pathogenesis and clinical features of disorders that cause traumatic hemolysis.
Hemolytic anemias are encountered less often than anemias due to decreased red cell production or blood loss. By far the most common hemolytic anemia in both pediatric and adult medicine is sickle cell disease (Chapter 9). Next in prevalence are acquired hemolytic anemias. Familiarity with the pathophysiology of this group of disorders is essential because these patients often pose formidable challenges in both diagnosis and management.
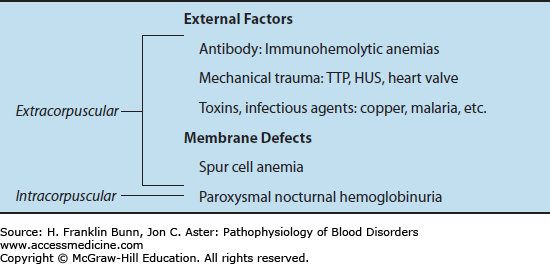
An overview of the hemolytic anemias is presented in Chapter 3 and summarized in Table 3-2. As indicated in Table 11-1, with the exception of the rare disorder paroxysmal nocturnal hemoglobinuria, all of the acquired hemolytic anemias are extracorpuscular. This means that red cells from a compatible donor are hemolyzed as readily as the patient’s own red cells. Acquired hemolysis is most often caused by immune-mediated destruction or traumatic mechanical damage of red cells.
IMMUNE HEMOLYTIC ANEMIAS
Immune hemolysis can be triggered by:
Autoantibodies binding to antigens on the patient’s red cells.
Alloantibodies binding to antigens on transfused red cells.
This chapter will deal with autoimmune hemolytic anemia. Alloimmune hemolysis will be covered in Chapter 25 (Transfusion Medicine).
Autoimmune hemolytic anemia is conveniently divided into two categories based on the temperature dependence of autoantibody binding to red cells, summarized in Table 11-2.
Warm autoantibodies bind avidly to the patient’s red cells at body temperature. They are immunoglobulin G (IgG) antibodies with specificity for Rh group antigens found on red cells of nearly all individuals. IgG-coated red cells are cleared primarily in the spleen, where they are engulfed by resident macrophages that have receptors for the constant region (Fc) of the heavy (H) chain of IgG. Figure 11-1A shows a macrophage from a patient with warm antibody hemolytic anemia that has just ingested two red cells and is about to destroy them. Alternatively, the patient’s IgG-coated red cells may bind to the surface of the macrophage but escape total engulfment. Under these circumstances, the macrophage nibbles at the red cell membrane and removes a small portion of it. As shown in Figure 11-1B, this depletion of the red cell surface area results in the formation of a spherocyte. Spherocytes are the predominant morphologic feature in two disorders: warm antibody autoimmune hemolytic anemia (Fig. 11-2A) and hereditary spherocytosis (Chapter 10). Even though these two types of anemia have very different pathogenic mechanisms, they produce identical morphologic alterations in red cells.
Patients with warm antibody immune hemolysis usually present with fatigue, shortness of breath, and general symptoms of anemia. Physical examination often reveals slight icterus and splenomegaly.
FIGURE 11-1
Conditioning of antibody-coated red cells by macrophages. A) Erythrophagocytosis of two red cells by a macrophage in a patient with immune hemolytic anemia. (Used with permission from Lichtman MA, Lichtman’s Atlas of Hematology. www.accessmedicine.com. McGraw-Hill, New York.) B) Conversion of a normal, disk-shaped red cell into a spherocyte by macrophage conditioning.
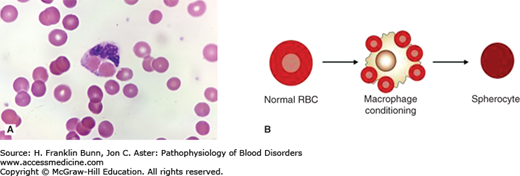
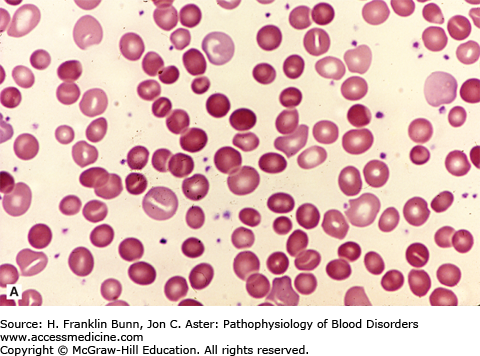
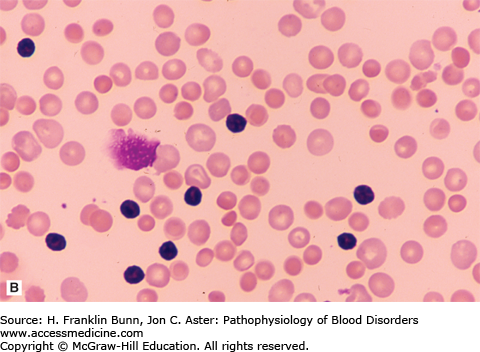
FIGURE 11-2
Blood films from patients with autoimmune hemolytic anemia due to a warm antibody. A) Idiopathic. B) Secondary to chronic lymphocytic leukemia. Note the presence of many spherocytes in both. (Used with permission from Lichtman MA, Lichtman’s Atlas of Hematology. www.accessmedicine.com. McGraw-Hill, New York, New York.)
Cold autoantibodies bind to the patient’s red cells with much higher affinity at low temperature than at body temperature. As a result, binding of cold autoantibodies to red cells is restricted to cool areas of the body, such as the extremities and the ear lobes. In the vast majority of cases, they are IgM macroglobulins that have specificity for the I antigen, which (like the Rh complex) is found on nearly all adult red cells. The large pentameric IgM binds the C3 component of complement much more efficiently than does IgG. As red cells return from the periphery and the blood warms, the IgM falls off the surface, but complement remains attached. C3-coated red cells are prey to two modes of destruction. If complement components are fully assembled, the cell will lyse and release its cytosolic contents into the plasma. More often, however, full fixation of complement does not occur, but sufficient C3b is deposited on the red cell surface for it to be recognized by C3 receptors on macrophages in the liver and elsewhere, which clear the cells from the circulation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


