Acid–Base Disorders
KEY CONCEPTS
![]() The kidney plays a central role in the regulation of acid–base homeostasis through the excretion or reabsorption of filtered bicarbonate (HCO3–), the excretion of metabolic fixed acids, and generation of new HCO3–.
The kidney plays a central role in the regulation of acid–base homeostasis through the excretion or reabsorption of filtered bicarbonate (HCO3–), the excretion of metabolic fixed acids, and generation of new HCO3–.
![]() Arterial blood gases (ABGs), along with serum electrolytes, physical findings, medical and medication history, and the clinical condition of the patient, are the primary tools to determine the cause of an acid–base disorder and to design and monitor a course of therapy.
Arterial blood gases (ABGs), along with serum electrolytes, physical findings, medical and medication history, and the clinical condition of the patient, are the primary tools to determine the cause of an acid–base disorder and to design and monitor a course of therapy.
![]() Metabolic acidosis and metabolic alkalosis are generated by a primary change in the serum bicarbonate concentration. In metabolic acidosis, bicarbonate is lost or a nonvolatile acid is gained, whereas metabolic alkalosis is characterized by a gain in bicarbonate or a loss of nonvolatile acid.
Metabolic acidosis and metabolic alkalosis are generated by a primary change in the serum bicarbonate concentration. In metabolic acidosis, bicarbonate is lost or a nonvolatile acid is gained, whereas metabolic alkalosis is characterized by a gain in bicarbonate or a loss of nonvolatile acid.
![]() Renal tubular acidosis (RTA) refers to a group of disorders characterized by impaired tubular renal acid handling despite normal or near-normal glomerular filtration rates. These patients often present with hyperchloremic metabolic acidosis.
Renal tubular acidosis (RTA) refers to a group of disorders characterized by impaired tubular renal acid handling despite normal or near-normal glomerular filtration rates. These patients often present with hyperchloremic metabolic acidosis.
![]() Respiratory compensation for a primary metabolic acidosis begins rapidly (within 15 to 30 minutes) but does not reach a steady state for 12 to 24 hours after the onset of metabolic acidosis.
Respiratory compensation for a primary metabolic acidosis begins rapidly (within 15 to 30 minutes) but does not reach a steady state for 12 to 24 hours after the onset of metabolic acidosis.
![]() Primary therapy of most acid–base disorders must include treatment or elimination of the underlying cause, not just correction of the pH and electrolyte disturbances.
Primary therapy of most acid–base disorders must include treatment or elimination of the underlying cause, not just correction of the pH and electrolyte disturbances.
![]() Potassium supplementation is always necessary for patients with chronic metabolic acidosis, as the bicarbonaturia resulting from alkali therapy increases the renal potassium wasting.
Potassium supplementation is always necessary for patients with chronic metabolic acidosis, as the bicarbonaturia resulting from alkali therapy increases the renal potassium wasting.
![]() Effective treatment of the underlying cause of some organic acidoses (e.g., ketoacidosis) can result in the regeneration of bicarbonate within hours, thus mitigating the need for alkali therapy.
Effective treatment of the underlying cause of some organic acidoses (e.g., ketoacidosis) can result in the regeneration of bicarbonate within hours, thus mitigating the need for alkali therapy.
![]() Loss of gastric acid from vomiting or nasogastric suctioning is often responsible for the development of a metabolic alkalosis, characterized by hypochloremia and hyperbicarbonatemia.
Loss of gastric acid from vomiting or nasogastric suctioning is often responsible for the development of a metabolic alkalosis, characterized by hypochloremia and hyperbicarbonatemia.
![]() Aggressive diuretic therapy can produce a metabolic alkalosis, and the accompanying hypokalemia can be serious.
Aggressive diuretic therapy can produce a metabolic alkalosis, and the accompanying hypokalemia can be serious.
![]() The patient’s response to volume replacement can be predicted by the urine chloride concentration and permits the differential diagnosis of metabolic alkalosis.
The patient’s response to volume replacement can be predicted by the urine chloride concentration and permits the differential diagnosis of metabolic alkalosis.
![]() Management of these disorders usually consists of treatment of the underlying cause of mineralocorticoid excess. In patients in whom the mineralocorticoid excess cannot be corrected, chronic pharmacologic therapy can be required.
Management of these disorders usually consists of treatment of the underlying cause of mineralocorticoid excess. In patients in whom the mineralocorticoid excess cannot be corrected, chronic pharmacologic therapy can be required.
![]() In most cases of acute metabolic acidosis, such as following cardiopulmonary arrest, sodium bicarbonate therapy is not indicated and can be detrimental. Blood gas analysis should guide therapy.
In most cases of acute metabolic acidosis, such as following cardiopulmonary arrest, sodium bicarbonate therapy is not indicated and can be detrimental. Blood gas analysis should guide therapy.
INTRODUCTION
Acid–base disorders are common and often serious disturbances that can result in significant morbidity and mortality. This chapter reviews the mechanisms responsible for the maintenance of acid–base balance and the laboratory analyses that aid clinicians in their assessment of acid–base disorders. The pathophysiology of the four primary acid–base disturbances is presented, evidence-based therapeutic options are reviewed, and management guidelines to optimize the outcome of patients with one of these disorders are presented. Given that medications are a frequent cause of acid–base abnormalities and that acid–base abnormalities are often preventable, clinicians must anticipate drug-related problems to avoid or minimize the clinical consequences of acid–base disorders, and when necessary, design appropriate treatment regimens.
ACID–BASE CHEMISTRY
An acid (in this equation, hydrochloric acid) is a substance that can donate protons (hydrogen ion [H+]):
(acid) HCl → H+ + chloride ion (Cl–)
A base (in this equation, ammonia [NH3]) is a substance that can accept protons (hydrogen ion [H+]):
Ammonia (NH3) + H+ → NH+4 (base)
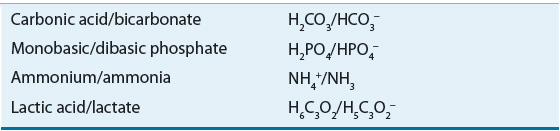
The acid–base pairs commonly encountered in clinical practice are listed in Table 37-1.
TABLE 37-1 Acid–Base Pairs
The acidity of body fluids is quantified in terms of the hydrogen ion concentration. By convention, the degree of acidity is expressed as pH, or the negative logarithm (base 10) of the hydrogen ion concentration. Thus, hydrogen ion concentration and pH are inversely related. Normally, the pH of blood is maintained at 7.40 ([H+] of 4 × 10–8 M) with a range of 7.35 to 7.45. A pH of less than 6.7 ([H+] of 2 × 10–7 M), representing a fivefold increase in hydrogen ion concentration, or greater than 7.7 ([H+] of 2 × 10–8 M), representing a 50% decrease in hydrogen ion concentration, is considered incompatible with life.
The hydrogen ion concentration in blood may not be indicative of that in other body compartments. For example, the pH within cells, within the cerebrospinal fluid, or on the surface of bone can all be altered without causing an alteration in blood pH.1 Recognizing this caveat, the acid–base status of the body is usually analyzed based on measurement of blood pH. Alterations in blood pH serve as the basis for the diagnosis of acid–base disorders.
Because the dissociation of acid–base pairs is an equilibrium reaction, the relationship between hydrogen ion concentration or pH and the relative concentrations of the acid and base can be described mathematically in terms of the dissociation constant for the acid–base buffer pair. When expressed as a logarithmic relationship, where pK is the negative logarithm of the dissociation constant K, this is known as the Henderson–Hasselbalch equation:
pH = pK + log([base]/[acid])
BUFFERS
The ability of a weak acid and its corresponding anion (base) to resist change in the pH of a solution on the addition of a strong acid or base is referred to as buffering. An acid–base pair is most efficient in functioning as a buffer at a pH close to its pK. The principal extracellular buffer is the carbonic acid/bicarbonate (H2CO3/HCO3–) system. Other physiologic buffers include plasma proteins, hemoglobin, and phosphates. Because the isohydric principle requires that all buffer systems remain in chemical equilibrium, the complex buffering of biologic fluids can be analyzed based on a single buffer pair.
The carbonic acid/bicarbonate buffer system plays a unique role in acid–base homeostasis. In addition to being the most abundant extracellular buffer, the components of this buffer pair are under dynamic regulation by the body. In the presence of carbonic anhydrase, carbonic acid, [H2CO3], is in equilibrium with carbon dioxide (CO2) gas. Changes in ventilation that alter the partial pressure of CO2 (PCO2) in the blood regulate the carbonic acid level in the blood. The bicarbonate concentration is independently regulated by the kidney. Because the pK for the carbonic acid/bicarbonate system is 6.1, the relationship between pH, carbonic acid, and bicarbonate concentrations can be described by the Henderson–Hasselbalch equation. The concentration of carbonic acid is directly proportional to the amount of CO2 dissolved in blood, which is equal to the product of PCO2 and its solubility in physiologic fluids (PCO2 × 0.03 for PCO2 expressed in mm Hg or PCO2 × 0.226 for PCO2 expressed in kPa). This term can, therefore, be substituted into the equation below in place of [H2CO3].
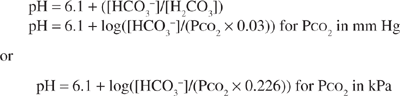
Thus, hydrogen ion concentration and pH are determined not by the absolute amounts of bicarbonate and PCO2, but by their ratio.1 Under normal physiologic conditions, the kidneys maintain the serum bicarbonate at approximately 24 mEq/L (24 mmol/L), whereas the lungs maintain the PCO2 at approximately 40 mm Hg (5.3 kPa). The normal physiologic pH is thus 7.4:
pH = 6.1 + log[24/(0.03 × 40)] (or pH = 6.1 + log[24/(0.226 × 5.3)])
pH = 6.1 + 1.3 = 7.4
If, in response to an acid load, the serum bicarbonate concentration were to decrease to 12 mEq/L (12 mmol/L), the predicted pH would be:
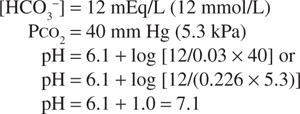
However, the normal respiratory response to an acid load is hyperventilation. As a result, if the PCO2 decreased to approximately 26 mm Hg (3.5 kPa), the change in pH would be less:
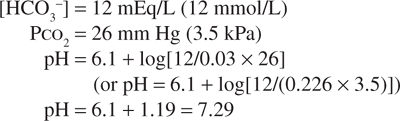
Thus, the physiologic regulation of both PCO2 and [HCO3–] permits the carbonic acid/bicarbonate system to provide more effective buffering of the extracellular fluids (ECFs) than could be achieved on the basis of chemical buffering alone.
REGULATION OF ACID–BASE HOMEOSTASIS
Cellular metabolism results in the production of large quantities of hydrogen that need to be excreted to maintain acid–base balance. In addition, small amounts of acid and alkali are also presented to the body through the diet. The bulk of acid production is in the form of CO2, with the average adult producing approximately 15,000 mmol of CO2 each day from the catabolism of carbohydrate, protein, and fat.2 When respiratory function is normal, the amount of CO2 produced metabolically is equal to the amount lost by respiration, and the blood CO2 concentration remains constant.
Digestion of dietary substances and tissue metabolism also result in the production of nonvolatile acids. These acids are derived primarily from the sulfur-containing amino acids cysteine and methionine, as well as from ingested sulfur. In addition, phosphates are generated from the metabolism of proteins and phospholipids. Neutral substances such as glucose can also be incompletely metabolized to intermediates, such as lactic and pyruvic acid, and fatty acids can be incompletely metabolized to acetoacetic acid and β-hydroxybutyric acid. These dietary and metabolic fixed acids are excreted, primarily by the kidney, to maintain acid–base homeostasis. On average, daily fixed acid excretion is approximately 0.8 mEq/kg per day (0.8 mmol/kg per day).3
Three mechanisms, each of which varies in its onset, collectively maintain acid–base balance: extracellular buffering, ventilatory regulation of carbon dioxide elimination, and renal regulation of hydrogen ion and bicarbonate excretion. Extracellular buffering occurs rapidly and is the body’s first defense against a sudden increase in hydrogen ion concentration. Hyperventilation will then result in a decrease in PCO2, returning blood pH toward normal. Finally, the kidney will excrete the excess hydrogen ion, with the resultant return of acid–base balance to normal over a period of day(s).
Extracellular Buffering
The body’s buffering system can be divided into three components: bicarbonate/carbonic acid, proteins, and phosphates. The bicarbonate buffer is the most important of the body’s buffers, because (a) there is more bicarbonate present in the ECF than any other buffer component; (b) the supply of carbon dioxide is unlimited; and (c) the acidity of ECF can be regulated by controlling either the bicarbonate concentration or the PCO2.
Carbonic acid represents the respiratory component of the buffer pair because its concentration is directly proportional to the PCO2, which is determined by ventilation. Bicarbonate represents the metabolic component because the kidney may alter its concentration by reabsorption, generating new bicarbonate, or altering elimination.4 The bicarbonate buffer system easily adapts to changes in acid–base status by alterations in ventilatory elimination of acid (PCO2) and/or renal elimination of base (HCO3–).
The phosphate buffer system consists of serum inorganic phosphate (3.5 to 5 mg/dL [1.13 to 1.62 mmol/L]), intracellular organic phosphate, and calcium phosphate in bone. Extracellular phosphate is present only in low concentrations, so its usefulness as a buffer is limited; however, as an intracellular buffer, phosphate is more useful. Calcium phosphate in bone is relatively inaccessible as a buffer, but prolonged metabolic acidosis will result in the release of phosphate from bone.
Intracellular and extracellular proteins also act as buffering systems. The charged side chains of amino acids provide the buffering action. Because the concentration of protein is much greater intracellularly than extracellularly, protein is much more important as an intracellular buffer.
Respiratory Regulation
The second mechanism for maintenance of acid–base homeostasis is control of ventilation. Both the rate and depth of ventilation can be varied to allow for excretion of CO2 generated by diet and tissue metabolism. Medullary chemoreceptors in the brainstem sense changes in PCO2 and in pH and modulate the control of breathing. Increasing minute ventilation (the total amount of air exhaled over a 1-minute period), by increasing respiratory rate and/or tidal volume (the amount of air exhaled in one breath), will increase CO2 excretion and decrease the blood PCO2. Conversely, decreasing minute ventilation decreases CO2 excretion and increases blood PCO2. This system rapidly adjusts within minutes to changes in acid–base balance.2
Renal Regulation
![]() Because bicarbonate is a small ion, it is freely filtered at the glomerulus. The bicarbonate load delivered to the nephron is approximately 4,500 mEq/day (4,500 mmol/day). To maintain acid–base balance, this entire filtered load must be reabsorbed. Bicarbonate reabsorption occurs primarily in the proximal tubule (Fig. 37-1). In the tubular lumen, filtered bicarbonate combines with hydrogen ion secreted by the apical sodium ion (Na+)–H+-exchanger to form carbonic acid. The carbonic acid is rapidly broken down to CO2 and water by carbonic anhydrase located on the luminal surface of the brush border membrane. The CO2 then diffuses into the proximal tubular cell, where it reforms carbonic acid in the presence of intracellular carbonic anhydrase. The carbonic acid dissociates to form hydrogen ions that can again be secreted into the tubular lumen, and bicarbonate that exits the cell across the basolateral membrane and enters the peritubular capillary.
Because bicarbonate is a small ion, it is freely filtered at the glomerulus. The bicarbonate load delivered to the nephron is approximately 4,500 mEq/day (4,500 mmol/day). To maintain acid–base balance, this entire filtered load must be reabsorbed. Bicarbonate reabsorption occurs primarily in the proximal tubule (Fig. 37-1). In the tubular lumen, filtered bicarbonate combines with hydrogen ion secreted by the apical sodium ion (Na+)–H+-exchanger to form carbonic acid. The carbonic acid is rapidly broken down to CO2 and water by carbonic anhydrase located on the luminal surface of the brush border membrane. The CO2 then diffuses into the proximal tubular cell, where it reforms carbonic acid in the presence of intracellular carbonic anhydrase. The carbonic acid dissociates to form hydrogen ions that can again be secreted into the tubular lumen, and bicarbonate that exits the cell across the basolateral membrane and enters the peritubular capillary.
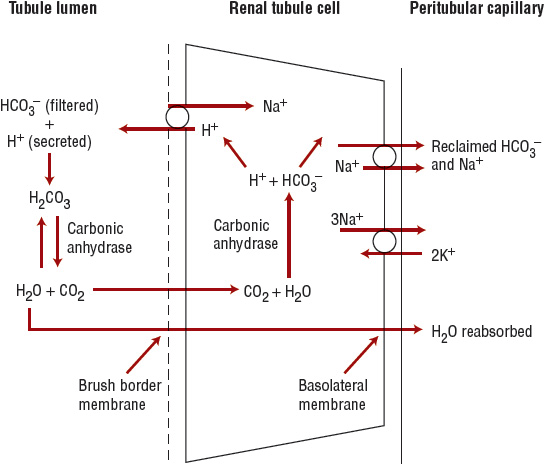
FIGURE 37-1 Proximal tubular bicarbonate reabsorption. In the tubular lumen, filtered bicarbonate (HCO3–) combines with hydrogen ion (H+) secreted by an apical sodium ion (Na+)–H+ exchanger to form carbonic acid (H2CO3). The carbonic acid is rapidly broken down to carbon dioxide (CO2) and water by carbonic anhydrase located on the luminal surface of the brush border membrane. The CO2 then diffuses into the proximal tubular cell, where it reforms carbonic acid in the presence of intracellular carbonic anhydrase. The carbonic acid dissociates the former hydrogen ion that can again be secreted into the tubular lumen, and bicarbonate that exits the cell across the basolateral membrane and enters the peritubular capillary.
Excretion of metabolic fixed acids and generation of new HCO3– is achieved through renal ammoniagenesis and distal tubular hydrogen ion secretion. Ammoniagenesis plays a critical role in acid–base homeostasis, with ammonium (NH4+) excretion comprising approximately 50% of renal net acid excretion. Ammonium is generated from the deamination of glutamine in the proximal tubule. For each ammonium ion excreted in the urine, one bicarbonate ion is regenerated and returned to the circulation.5
Distal tubular hydrogen ion secretion accounts for the remaining 50% of net acid excretion (Fig. 37-2). In the distal tubular cell, CO2 combines with water in the presence of intracellular carbonic anhydrase to form carbonic acid, which dissociates to H+ and HCO3–. The H+ is actively transported into the tubular lumen by a H+–adenosine triphosphatase (ATPase). The bicarbonate exits the cell across the basolateral membrane and enters the circulation.5
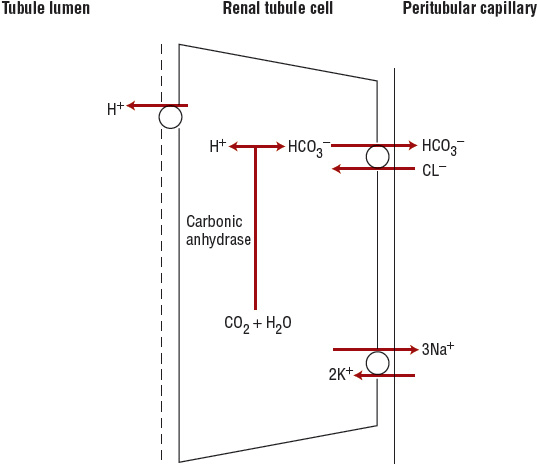
FIGURE 37-2 Collecting duct acid excretion. Hydrogen ion (H+) and bicarbonate (HCO3–) are generated intracellularly from carbon dioxide (CO2) and water, in the presence of intracellular carbonic anhydrase. The hydrogen ion is actively secreted into the tubular lumen by H+–ATPase located in the apical (luminal) membrane. Bicarbonate exits the cell across the basolateral membrane and enters the peritubular capillary. (Cl–, chloride ion; Na+, sodium ion.)
ACID–BASE DISTURBANCES
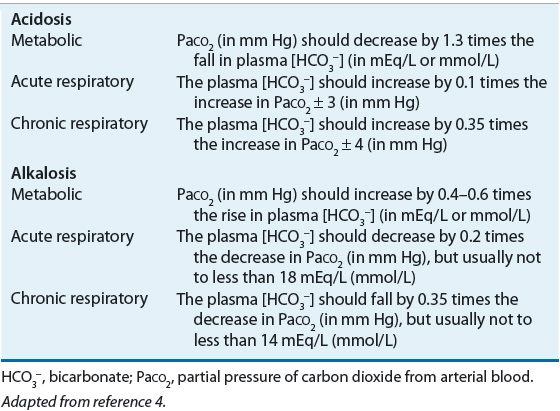
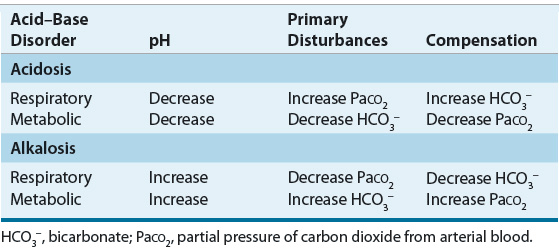
![]() Alterations in blood pH are designated by the suffix “-emia”; acidemia is an arterial blood pH <7.35 and alkalemia is an arterial blood pH >7.45. The pathophysiologic processes that result in alterations in blood pH are designated by the suffix “-osis.” These disturbances are classified as either metabolic or respiratory in origin. In metabolic acid–base disorders, the primary disturbance is in the plasma bicarbonate concentration. Metabolic acidosis is characterized by a decrease in the plasma bicarbonate concentration whereas in metabolic alkalosis the plasma bicarbonate concentration is increased. Respiratory acid–base disorders are caused by alterations in alveolar ventilation that produce corresponding changes in the partial pressure of carbon dioxide from arterial blood (PaCO2). In respiratory acidosis, the PaCO2 is elevated; in respiratory alkalosis, it is decreased. Each disturbance has a compensatory (secondary) response that attempts to correct the HCO3–-to-PaCO2 ratio toward normal and mitigate the change in pH (Table 37-2). Although the time course of the respiratory compensatory responses to metabolic disturbances is rapid, the metabolic compensation for respiratory disturbances is slow. As a result, respiratory disturbances are characterized as acute (minutes to hours in duration), indicating that there has not been sufficient time for metabolic compensation, or chronic (days), indicating that sufficient time for metabolic compensation has elapsed.
Alterations in blood pH are designated by the suffix “-emia”; acidemia is an arterial blood pH <7.35 and alkalemia is an arterial blood pH >7.45. The pathophysiologic processes that result in alterations in blood pH are designated by the suffix “-osis.” These disturbances are classified as either metabolic or respiratory in origin. In metabolic acid–base disorders, the primary disturbance is in the plasma bicarbonate concentration. Metabolic acidosis is characterized by a decrease in the plasma bicarbonate concentration whereas in metabolic alkalosis the plasma bicarbonate concentration is increased. Respiratory acid–base disorders are caused by alterations in alveolar ventilation that produce corresponding changes in the partial pressure of carbon dioxide from arterial blood (PaCO2). In respiratory acidosis, the PaCO2 is elevated; in respiratory alkalosis, it is decreased. Each disturbance has a compensatory (secondary) response that attempts to correct the HCO3–-to-PaCO2 ratio toward normal and mitigate the change in pH (Table 37-2). Although the time course of the respiratory compensatory responses to metabolic disturbances is rapid, the metabolic compensation for respiratory disturbances is slow. As a result, respiratory disturbances are characterized as acute (minutes to hours in duration), indicating that there has not been sufficient time for metabolic compensation, or chronic (days), indicating that sufficient time for metabolic compensation has elapsed.
TABLE 37-2 Interpretation of Simple Acid–Base Disorders
CLINICAL ASSESSMENT OF ACID–BASE STATUS
![]() Blood gases are measured to determine the patient’s oxygenation and acid–base status. Under normal circumstances, there is no clinically significant difference in pH between arterial and mixed venous blood. Arterial samples are designated with the letter “a” (e.g., partial pressure of oxygen from arterial blood [PaO2] and PaCO2), whereas mixed venous samples are labeled with the letter “v” or not labeled (e.g., partial pressure of oxygen from venous blood [PvO2] and partial pressure of carbon dioxide from venous blood [PvCO2]). The normal values for arterial and venous blood gases are shown in Table 37-3. Arterial blood reflects how well the blood is being oxygenated by the lungs (an accurate measurement of PaO2), whereas venous blood reflects how much oxygen tissues are using. Arterial blood rather than venous blood should be used whenever possible because venous blood obtained from an extremity can provide misleading information. If metabolism in the extremity is altered by hypoperfusion, exercise, infection, or some other cause, the difference in the amount of dissolved oxygen between arterial and venous blood can be dramatic. The venous pH and PCO2 during cardiopulmonary resuscitation might be significantly lower and higher, respectively, than the arterial pH and arterial PCO2. This indicates a severe tissue acidosis from CO2 accumulation caused by hypoperfusion.
Blood gases are measured to determine the patient’s oxygenation and acid–base status. Under normal circumstances, there is no clinically significant difference in pH between arterial and mixed venous blood. Arterial samples are designated with the letter “a” (e.g., partial pressure of oxygen from arterial blood [PaO2] and PaCO2), whereas mixed venous samples are labeled with the letter “v” or not labeled (e.g., partial pressure of oxygen from venous blood [PvO2] and partial pressure of carbon dioxide from venous blood [PvCO2]). The normal values for arterial and venous blood gases are shown in Table 37-3. Arterial blood reflects how well the blood is being oxygenated by the lungs (an accurate measurement of PaO2), whereas venous blood reflects how much oxygen tissues are using. Arterial blood rather than venous blood should be used whenever possible because venous blood obtained from an extremity can provide misleading information. If metabolism in the extremity is altered by hypoperfusion, exercise, infection, or some other cause, the difference in the amount of dissolved oxygen between arterial and venous blood can be dramatic. The venous pH and PCO2 during cardiopulmonary resuscitation might be significantly lower and higher, respectively, than the arterial pH and arterial PCO2. This indicates a severe tissue acidosis from CO2 accumulation caused by hypoperfusion.
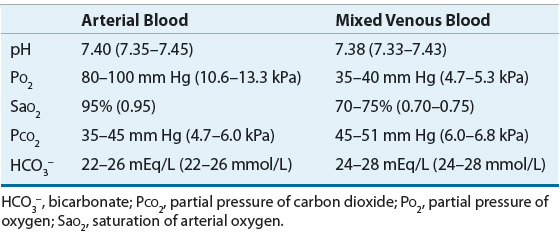
TABLE 37-3 Normal Blood Gas Values
Analysis of Arterial Blood Gas Data
ABGs provide an assessment of the patient’s acid–base status.6 Low pH values (<7.35) indicate an acidemia, whereas high pH values (>7.45) indicate an alkalemia (Fig. 37-3). In a metabolic acidosis, the pH is decreased in association with a decreased serum bicarbonate concentration and a compensatory decrease in PaCO2. In a respiratory acidosis, the pH is decreased; the PaCO2, however, is elevated. The serum bicarbonate concentration is variable, depending on whether it is an acute disturbance (minimal increase in serum bicarbonate) or a chronic respiratory acidosis (substantial increase in serum bicarbonate). In a metabolic alkalosis, the pH is elevated in association with an increased bicarbonate concentration and a compensatory increase in PaCO2. In a respiratory alkalosis, the pH is also elevated; the PaCO2, however, is decreased. As with respiratory acidosis, the metabolic compensation is variable: a minimal decrease in serum bicarbonate is often noted in acute respiratory alkalosis while a larger decrease in [HCO3–] is common with chronic respiratory alkalosis. Although each measurement has a normal range (see Table 37-3), it is often easiest to consider the midpoint of each range as the normal value. This would correlate to a pH of 7.4, PaCO2 of 40 mm Hg (5.3 kPa), and HCO3– of 24 mEq/L (24 mmol/L). Steps in acid–base interpretation are described in Table 37-4.
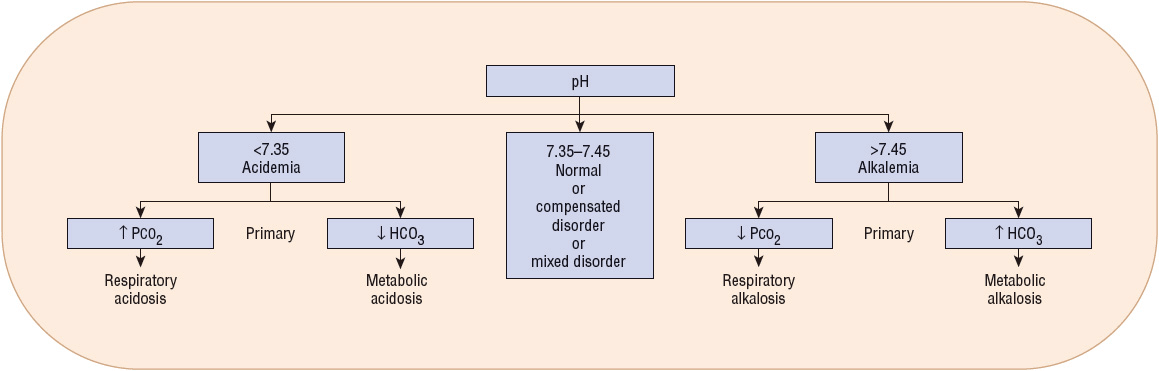
FIGURE 37-3 Analysis of arterial blood gases. (HCO3–, bicarbonate; PCO2, partial pressure of carbon dioxide.)
TABLE 37-4 Steps in Acid–Base Diagnosis

When ABGs differ significantly from those expected on the basis of the patient’s clinical condition and previous laboratory determinations, additional venous blood samples should be drawn to assess plasma electrolyte concentrations. The bicarbonate calculated from the patient’s PaCO2 and pH of the blood gas should be compared with the measured total CO2 content (the amount of CO2 gas extractable from plasma, consisting of HCO3–, H2CO3, and PCO2). Ordinarily, the blood gas bicarbonate value is approximately 1 to 2 mEq/L (1 to 2 mmol/L) less than total CO2 content.3 If these values do not correspond, the results should be interpreted with caution because the difference can reflect an error in the blood collection or storage of the sample, or in the calibration of the blood gas analyzer.
METABOLIC ACID–BASE DISORDERS
Metabolic Acidosis
Metabolic acidosis is characterized by a decrease in pH as the result of a primary decrease in serum bicarbonate concentration.
Pathophysiology
Metabolic acidosis can result from the buffering (consumption of HCO3–) of an exogenous acid, an organic acid accumulating because of a metabolic disturbance (e.g., lactic acid or ketoacids), or the progressive accumulation of endogenous acids secondary to impaired renal function (e.g., phosphates and sulfates).7 The serum HCO3– can also be decreased as the result of a loss of bicarbonate-rich body fluids (e.g., diarrhea, biliary drainage, or pancreatic fistula) or occur secondary to the rapid administration of non-alkali–containing IV fluids (dilutional acidosis).8
The serum anion gap (SAG), as defined below, can be used to infer whether an organic or mineral acidosis is present.
![]()
To maintain electroneutrality, the total concentration of cations in the serum must equal the total concentration of anions.
![]()
The cation concentration is equal to the sodium concentration plus that of “unmeasured” cations (UCs), predominantly magnesium, calcium, and potassium. The anion concentration is equal to the concentrations of chloride, bicarbonate, and “unmeasured” anions (UAs), including proteins, sulfates, phosphates, and organic anions. Therefore, as the result of the combination of the two equations above, the SAG can be expressed as:
![]()
The normal SAG is approximately 9 mEq/L (9 mmol/L), with a range of 3 to 11 mEq/L (3 to 11 mmol/L). This value is lower than the value of 12 mEq/L (12 mmol/L) cited in the literature in the past because of changes in the instrumentation for measurement of serum electrolytes.7 Increases in the anion gap (AG) to values in excess of 17 to 20 mEq/L (17 to 20 mmol/L) are indicative of the accumulation of unmeasured anions in ECF.
These unmeasured anions are generated as the result of the consumption of HCO3– by endogenous organic acids such as lactic acid, acetoacetic acid, or β-hydroxybutyric acid or from the ingestion of toxins such as methanol or ethylene glycol. The degree of elevation in the SAG is dependent on the clearance of the anion, as well as the multiple factors that influence HCO3– concentrations. Thus, the SAG is a relative rather than an absolute indication of the cause of metabolic acidosis. The SAG can also be elevated in the metabolic acidosis because of renal failure, as the result of the accumulation of various organic anions, phosphates, and sulfates.
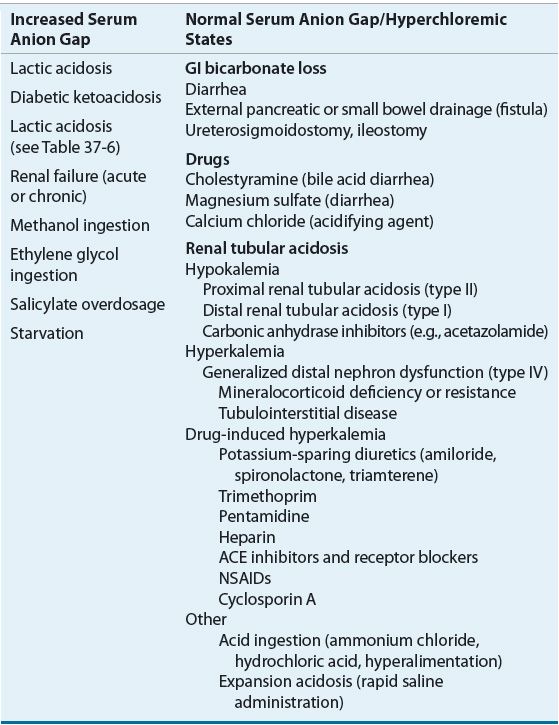
In hyperchloremic metabolic acidosis, bicarbonate losses from the ECF are replaced by chloride, and the SAG remains normal. This decrease in bicarbonate may be due to GI tract losses, dilution of bicarbonate in the ECF as the result of the addition of sodium chloride solutions or chloride-containing acids. Common causes of metabolic acidosis with an increased or a normal SAG are listed in Table 37-5.
TABLE 37-5 Common Causes of Metabolic Acidosis
Hyperchloremic Metabolic Acidosis
Hyperchloremic metabolic acidosis can result from increased GI bicarbonate loss, renal bicarbonate wasting, impaired renal acid excretion, or exogenous acid gain. GI disorders such as diarrhea, biliary, or pancreatic drainage through either a surgical drain or fistula can result in the loss of large volumes of bicarbonate-containing fluids. Severe diarrhea, the most common cause of hyperchloremic metabolic acidosis, can lead to a daily loss of 5 to 10 L of fluid containing 100 to 140 mEq/L (100 to 140 mmol/L) of sodium, 20 to 40 mEq/L (20 to 40 mmol/L) of potassium, 80 to 100 mEq/L (80 to 100 mmol/L) of chloride, and 30 to 50 mEq/L (30 to 50 mmol/L) of bicarbonate.4 Patients who have undergone ureteral diversion into the sigmoid colon or isolated ileal loop can also develop a hyperchloremic metabolic acidosis. This is the result of a net loss of bicarbonate given that chloride is reabsorbed and bicarbonate is secreted by GI epithelial cells in the presence of the urine that is retained in the colon or bowel loop.
Hyperchloremic metabolic acidosis caused by renal bicarbonate wasting is the defining disturbance in proximal RTA and is a complication of therapy with carbonic anhydrase inhibitors, particularly when they are administered for more than 24 to 48 hours. During the treatment of diabetic ketoacidosis, renal loss of β-hydroxybutyrate and acetoacetate, which would otherwise be metabolized to yield bicarbonate, can contribute to the development of hyperchloremic metabolic acidosis. Impaired renal acid excretion that occurs as a result of distal tubular dysfunction in patients with distal RTAs can also occur in patients with moderate to severe renal insufficiency from other causes. The metabolic acidosis of renal insufficiency is initially hyperchloremic but can progress to an anion-gap acidosis as the renal insufficiency worsens and sulfates, phosphates, and other anions accumulate. Hyperchloremic metabolic acidosis can also result from the exogenous administration of acid (hydrochloric acid, ammonium chloride) or the unbuffered administration of acid salts from the amino acids in total parenteral nutrition fluids.
Renal Tubular Acidosis
![]() Renal tubular disorders can involve the proximal tubule, with a resultant failure to reabsorb filtered bicarbonate, or affect acid excretion in the distal tubule. The distal RTAs are the most common, and are all characterized by impaired net acid excretion. The distal RTAs are subdivided into those that are associated with hypokalemia (type I) and those associated with hyperkalemia (type IV). Patients with classic distal (type I) RTA have impaired hydrogen ion secretion and are unable to excrete the daily acid load necessary to maintain acid–base balance.4 These patients are unable to maximally acidify their urine (i.e., attain urine pH <5.5), even in the face of an acid challenge. Type I RTA may be the result of a primary tubular defect or develop secondary to a wide variety of disorders including hypercalcemia, multiple myeloma, systemic lupus erythematosus, Sjögren’s syndrome, sickle-cell disease, and renal transplant rejection, or following the administration of amphotericin B or ingestion of toluene. The primary form of this disorder usually occurs in children and can result in severe acidosis, slowed growth, nephrocalcinosis, and kidney stones.7,9 In adults, clinical complications include osteomalacia, nephrocalcinosis, and recurrent kidney stones. The hypokalemia associated with classic distal (type I) RTA results from secondary hypoaldosteronism associated with volume depletion. The renal potassium wasting decreases considerably if bicarbonate therapy is administered.
Renal tubular disorders can involve the proximal tubule, with a resultant failure to reabsorb filtered bicarbonate, or affect acid excretion in the distal tubule. The distal RTAs are the most common, and are all characterized by impaired net acid excretion. The distal RTAs are subdivided into those that are associated with hypokalemia (type I) and those associated with hyperkalemia (type IV). Patients with classic distal (type I) RTA have impaired hydrogen ion secretion and are unable to excrete the daily acid load necessary to maintain acid–base balance.4 These patients are unable to maximally acidify their urine (i.e., attain urine pH <5.5), even in the face of an acid challenge. Type I RTA may be the result of a primary tubular defect or develop secondary to a wide variety of disorders including hypercalcemia, multiple myeloma, systemic lupus erythematosus, Sjögren’s syndrome, sickle-cell disease, and renal transplant rejection, or following the administration of amphotericin B or ingestion of toluene. The primary form of this disorder usually occurs in children and can result in severe acidosis, slowed growth, nephrocalcinosis, and kidney stones.7,9 In adults, clinical complications include osteomalacia, nephrocalcinosis, and recurrent kidney stones. The hypokalemia associated with classic distal (type I) RTA results from secondary hypoaldosteronism associated with volume depletion. The renal potassium wasting decreases considerably if bicarbonate therapy is administered.
The hyperkalemic distal (type IV) RTAs are a heterogeneous group of disorders characterized by hypoaldosteronism or generalized distal tubule defects. The most common form of type IV RTA is hyporeninemic hypoaldosteronism. This syndrome is most commonly associated with mild renal insufficiency caused by diabetic nephropathy, but can also be seen in a variety of other disorders, including chronic interstitial nephritis, sickle-cell disease, human immunodeficiency virus (HIV) nephropathy, and obstructive uropathy. The clinical presentation of this syndrome is often exacerbated by pharmacologic therapy with agents that can interfere with the renin–angiotensin–aldosterone axis, such as β-adrenergic blockers, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, and nonsteroidal antiinflammatory drugs (NSAIDs). Heparin can induce the syndrome by inhibiting adrenal aldosterone biosynthesis. Patients with this form of RTA are able to maximally acidify their urine (urine pH <5.5).9 The primary defect in acid excretion is impaired ammoniagenesis caused by mild renal insufficiency. Hyperaldosteronism predisposes to the development of hyperkalemia, which results in further impairment of ammoniagenesis. Treatment to control the hyperkalemia is usually sufficient to reverse the metabolic acidosis, and mineralocorticoid replacement is frequently unnecessary.
Hyperkalemic distal (type IV) RTA resulting from generalized distal tubule defects is less common than hyporeninemic hypoaldosteronism but is more common than classic distal (type I) RTA. Patients with this defect have impaired tubular potassium secretion in addition to impaired urinary acidification (urine pH >5.5, despite acidemia or acid loading). Urinary obstruction is the most frequent cause of this disorder, which can also be associated with sickle-cell nephropathy, systemic lupus erythematosus, HIV nephropathy, analgesic abuse nephropathy, amyloidosis, renal transplant rejection, and chronic cyclosporine nephrotoxicity.
Proximal (type II) RTA is characterized by defects in proximal tubular reabsorption of bicarbonate. Normally, more than 85% of filtered bicarbonate is reabsorbed in the proximal tubule. Defects in proximal tubular bicarbonate reabsorption result in increased delivery of bicarbonate to the distal nephron, which has a limited capacity for bicarbonate reabsorption. As a result, at a normal serum bicarbonate concentration, the filtered bicarbonate load is incompletely reabsorbed, and is lost in the urine. As the serum bicarbonate concentration decreases, the filtered load of bicarbonate is proportionately decreased. A new equilibrium is established in which the kidney is able to reabsorb the filtered bicarbonate load, albeit at a reduced serum bicarbonate concentration. Thus, patients with proximal RTA present with a chronic, nonprogressive hyperchloremic metabolic acidosis. These patients are able to acidify their urine in response to an acid load, but develop bicarbonaturia at a reduced serum bicarbonate concentration following bicarbonate loading. The impaired bicarbonate reabsorption results in salt wasting and secondary hyperaldosteronism. Hypokalemia, which can be severe, usually develops as a result of the hyperaldosteronism and bicarbonaturia.4,7 Unlike patients with classic distal (type I) RTA, the hyperkalemia if present in proximal RTA is exacerbated by alkali replacement. Proximal RTA can develop as an isolated defect, or it can be associated with generalized proximal tubular dysfunction (Fanconi’s syndrome), with impaired proximal tubular glucose, phosphate, and amino acid reabsorption. Proximal RTA usually presents as an acquired disorder, secondary to a variety of diseases (amyloidosis, multiple myeloma, or nephrotic syndrome) or exposure to toxins (lead, cadmium, mercury, or outdated tetracyclines). Pharmacologic therapy with carbonic anhydrase inhibitors produces an iatrogenic form of proximal RTA.
Elevated Anion Gap Metabolic Acidosis
Metabolic acidosis with an increased SAG commonly results from increased endogenous organic acid production.7 In lactic acidosis, lactic acid accumulates as a by-product of anaerobic metabolism. Accumulation of the ketoacids β-hydroxybutyric acid and acetoacetic acid defines the ketoacidosis of uncontrolled diabetes mellitus, alcohol intoxication, and starvation (see Table 37-5). In advanced renal failure, accumulation of phosphate, sulfate, and organic anions is responsible for the increased SAG, which is usually less than 24 mEq/L (24 mmol/L).7 The severe metabolic acidosis seen in myoglobinuric acute renal failure caused by rhabdomyolysis may be caused by the metabolism of large amounts of sulfur-containing amino acids released from myoglobin.
The presence of mild elevations in the SAG cannot be automatically attributed to the presence of a high SAG metabolic acidosis. Elevations in the SAG are commonly seen in hospitalized patients, especially those who are critically ill.7,10 A variety of factors can contribute to this nonspecific elevation in the SAG, including the presence of alkalemia, which increases the anionic charge of albumin and other plasma proteins. The usefulness of the SAG as a marker of acid–base status is dependent on proper interpretation of a patient’s clinical status.5,11 Despite these limitations, when the SAG exceeds 20 to 25 mEq/L (20 to 25 mmol/L) a significant organic acidosis is likely to be present.
High anion gap metabolic acidosis can develop in many clinical settings, including uncontrolled diabetes mellitus (see Chap. 57), alcohol intoxication (see Chaps. 24 and 49), and starvation (see Chap. 47). Toxic ingestions of methanol and ethylene glycol are also associated with high anion gap metabolic acidosis and can be differentiated from other causes of SAG because of the presence of an elevated osmolar gap. The mechanisms responsible for the development of acidosis in these settings are diverse.7,12
Lactic Acidosis Lactic acidosis is one of the most common causes of high SAG metabolic acidosis and can impact approximately 1% of hospitalized patients. Lactic acid is the end product of anaerobic metabolism of glucose (glycolysis).7 In normal individuals, lactic acid derived from pyruvate enters the circulation in small amounts and is promptly removed by the liver. In the liver, and to a lesser extent in the kidney, lactic acid is reoxidized to pyruvic acid, which is then metabolized to CO2 and H2O. The normal plasma lactate concentration in healthy subjects is approximately 1 mEq/L (1 mmol/L).3,7,12 The diagnosis of lactic acidosis should be considered in all patients with metabolic acidosis associated with an increased SAG. Lactic acidosis is considered to be present when lactate concentrations exceed 4 to 5 mEq/L (4 to 5 mmol/L) in an acidemic patient.
Classically, lactic acidosis has been differentiated into disorders associated with tissue hypoxia (type A lactic acidosis) and disorders associated with deranged oxidative metabolism (type B lactic acidosis), although the distinction between them is blurred (Table 37-6).7,11,12 The etiologies of lactic acidosis can also be categorized on the basis of changes in lactate production and/or utilization.7,12 Metabolic disturbances can result in increased tissue pyruvate production or impaired utilization, with proportional increases in lactate concentrations. Increased lactate production is more commonly associated with alterations in tissue redox state, resulting in preferential conversion of pyruvate to lactate. During anaerobic metabolism, reduced nicotinamide adenine dinucleotide accumulates, driving the conversion of pyruvate to lactate and increasing the lactate-to-pyruvate ratio. States of enhanced metabolic activity (e.g., grand mal seizures, strenuous exercise, or hyperthermia), decreased tissue oxygen delivery (e.g., severe anemia, hypoxia, circulatory shock, or carbon monoxide poisoning), or impaired oxygen utilization (e.g., cyanide toxicity) all are associated with lactic acidosis. Impaired hepatic clearance of lactate, as seen in hypoperfusion states, liver failure, and alcohol intoxication, can also result in lactic acidosis.
TABLE 37-6 Causes of Lactic Acidosis
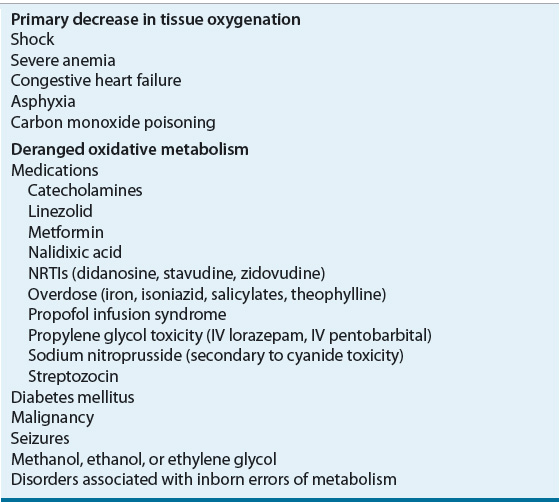
Cardiovascular and septic shock, with resultant tissue hypoperfusion, are the most common causes of lactic acidosis. Poor tissue perfusion and hypoxia influence enzymatic pyruvate and lactate metabolism to stimulate anaerobic glycolysis and to decrease lactate utilization. This leads to hyperlactatemia and lactic acidosis. The mortality rate of this type of lactic acidosis can be as high as 80% and correlates with the degree of hyperlactatemia.
Lactic acidosis associated with liver disease, toxins, and congenital enzyme deficiency can be caused by deranged oxidative metabolism or impaired lactate clearance.4,7,12 The exact role of diabetes mellitus in the induction of lactic acidosis is not clear. It may involve a decrease in pyruvate dehydrogenase activity, the enzyme responsible for pyruvate metabolism. Lactic acidosis in neoplastic disease is uncommon and reported mostly in patients with myeloproliferative disorders. Leukocytes and neoplastic cells in general have high rates of glycolysis. In the case of a large tumor or tightly packed bone marrow, oxygenation can be decreased, favoring the accumulation of lactate. Lactic acidosis has been reported in patients with massive liver tumors, and it has been postulated that the liver uptake of lactate is decreased in these patients. Lactic acidosis associated with seizures is usually transient and occurs because of excessive muscle activity.13
A number of medications can cause lactic acidosis.13–23 Two of the most common medications associated with the development of lactic acidosis are nucleoside-analog reverse transcriptase inhibitors (NRTIs) (3.9 cases per 1,000 person-years) and metformin (0.03 cases per 1,000 person-years).15–17 The proposed mechanism of NRTI-induced lactic acidosis is the inhibition of the enzyme DNA polymerase gamma that is responsible for mitochondrial DNA synthesis.15 Disruption of this enzyme can inhibit the transport of lactate into the mitochondria, leading to an accumulation in the cytoplasm. Stavudine is the NRTI most frequently associated with lactic acidosis; however, the combination of stavudine and didanosine confers the highest risk.
The primary suspected mechanism for metformin-induced lactic acidosis is inhibition of liver gluconeogenesis as the result of its inhibitory effects on pyruvate carboxylase which is necessary for the conversion of pyruvate to glucose.16,17 Other possible pathways for metformin-associated lactic acidosis include a decrease in both hepatic intracellular pH and cardiac output and an increase in lactate production in the gut and increased renal loss of bicarbonate.13 Risk factors for metformin-induced lactic acidosis include renal insufficiency, liver disease, dehydration, advanced age, alcohol consumption, and supratherapeutic dosing. Metformin should be discontinued during periods of tissue hypoxia (e.g., myocardial infarction, sepsis), for 3 days after contrast media has been administered or 2 days before general anesthesia administration. In the latter two cases, metformin should only be reinstituted when the patient’s renal function is stable.
Propylene glycol is commonly used as a solubilizing agent in IV drug preparations (e.g., lorazepam, pentobarbital) and is predominantly metabolized to lactic acid via the hepatic enzyme alcohol dehydrogenase. The administration of large doses of propylene glycol, particularly to patients with renal or liver insufficiency, can lead to a lactic acidosis with an osmolar gap and thus serial measurement of the osmolar gap can be used to detect propylene glycol accumulation.18,19
Reports of the association between propofol and lactic acidosis were initially described in children.20 This association is now recognized in adults and has come to be known as the propofol-related infusion syndrome. In addition to lactic acidosis, cardiac failure, rhabdomyolysis, and renal failure have been observed primarily because of uncoupling of oxidative phosphorylation and impaired oxidation of free fatty acids. This syndrome is most frequently seen in patients receiving propofol at high doses (>5 mg/kg/h) for more than 2 days.
Clinical Presentation
Chronic metabolic acidosis is usually not associated with severe acidemia and is relatively asymptomatic. The major manifestations are in the bones, where chronic acidemia causes bone demineralization with the development of rickets in children and osteomalacia and osteopenia in adults.26 In infants and children, chronic metabolic acidosis is associated with growth failure and short stature and can be associated with nonspecific symptoms including anorexia, nausea, weight loss, and muscle weakness.
Severe metabolic acidosis is usually associated with acute processes. The manifestations of severe acidemia (pH <7.20) involve the cardiovascular, respiratory, and CNS. Hyperventilation is often the first sign of metabolic acidosis. At a pH of 7.2, pulmonary ventilation increases approximately fourfold, and an eightfold increase has been noted at a pH of 7.24,25 Respiratory compensation can occur as Kussmaul respirations—the deep, rapid respirations seen commonly in patients with diabetic ketoacidosis. In extremely severe acidosis (pH <6.8), CNS function is disrupted to such a degree that the respiratory center is depressed.
CNS depression correlates more closely with spinal fluid pH than with blood pH. For this reason, neurologic symptoms tend to occur more frequently and to a greater degree in patients with respiratory acidosis because the CO2 accumulated in the respiratory form readily crosses the blood–brain barrier to cause acidosis in the CNS.1 Because of the slow penetration of administered bicarbonate into the CNS, the CNS pH fails to normalize as rapidly as blood pH. Therefore patients continue to hyperventilate because of sustained CNS acidity, and severe respiratory alkalosis can occur. Sustained lowering of the PaCO2 within 12 to 36 hours is to be anticipated during the correction of any metabolic acidosis.1
Systemic acidosis can cause peripheral arteriolar dilatation, characterized by flushing, a rapid heart rate, and wide pulse pressure. Initially, cardiac output can be increased, but as acidosis becomes more severe, myocardial contractility becomes impaired, and cardiac output decreases. The effects of vagal stimulation are also enhanced at pH levels lower than 7.1, probably as a consequence of inhibition of acetylcholinesterase. This increases the danger of vagally mediated bradycardia and heart block during acidosis.
GI symptoms of metabolic acidosis include loss of appetite, nausea, and vomiting. Severe acidosis (pH <7.1) interferes with carbohydrate metabolism and insulin utilization, and results in hyperglycemia. Metabolic acidosis alters potassium homeostasis and contributes to the development of hyperkalemia. The magnitude of the effect on serum potassium depends on the type of acidosis: Acidosis caused by mineral acids (e.g., hydrochloric acid) is associated with a greater change in potassium levels than acidosis caused by organic acids (e.g., lactic acidosis), in which the increase in potassium attributable to the acidosis per se is minimal.
Compensation
![]() The patient’s primary means to compensate for metabolic acidosis is to increase carbon dioxide excretion by increasing the respiratory rate. This results in a decrease in PaCO2. This ventilatory compensation results from stimulation of the respiratory center by changes in cerebral bicarbonate concentration and pH.1 For every 1-mEq/L (1 mmol/L) decrease in bicarbonate concentration below the average of 24, the PaCO2 decreases by approximately 1 to 1.5 mm Hg (0.13 to 0.20 kPa) from the normal value of 40 (5.3 kPa) (Table 37-7).
The patient’s primary means to compensate for metabolic acidosis is to increase carbon dioxide excretion by increasing the respiratory rate. This results in a decrease in PaCO2. This ventilatory compensation results from stimulation of the respiratory center by changes in cerebral bicarbonate concentration and pH.1 For every 1-mEq/L (1 mmol/L) decrease in bicarbonate concentration below the average of 24, the PaCO2 decreases by approximately 1 to 1.5 mm Hg (0.13 to 0.20 kPa) from the normal value of 40 (5.3 kPa) (Table 37-7).
TABLE 37-7 Guidelines for Initial Interpretation of Acid–Base Disorders