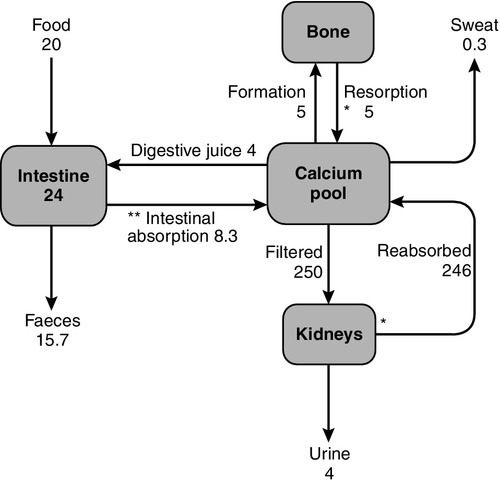
CHAPTER 6 Timothy Cundy; Andrew Grey; Ian R. Reid CHAPTER OUTLINE Regulation of calcium metabolism Biochemical assessment of calcium metabolism Distribution of body phosphorus Calcium is a divalent cation with multiple roles in vertebrate physiology, which can be grouped as either structural or metabolic. Its structural role is in the skeleton where calcium deficiency leads to skeletal disease, either in the form of osteoporosis or osteomalacia. Its metabolic roles are more numerous. The extracellular concentration of calcium ions influences the threshold for nerve action potentials, a high calcium raising the threshold and a low calcium having the opposite effect. The extracellular calcium concentration appears to alter the gating of sodium and potassium channels, possibly the result of calcium ions being attracted to and ‘screening’ the negative charge on the cell surface in the region of these channels. Calcium has a key role in intracellular signalling. The depolarization of nerve or muscle cells, or the binding of a hormone or cytokine to its receptor on the surface of other types of cell, results in an increase in cytosolic calcium concentration via either one or both of two mechanisms: the influx of calcium through plasma membrane channels and the release of calcium from intracellular stores (e.g. the sarcoplasmic or endoplasmic reticulum). The latter pathway depends on receptor-activated hydrolysis of inositol phospholipids in the plasma membrane, leading to the release of inositol trisphosphate into the cytosol. This compound binds to a receptor on the endoplasmic reticulum resulting in the release of calcium into the cytosol. The regulation of a variety of cell functions can then follow, some enzymes being directly affected by the cytosolic calcium concentration (e.g. protein kinase C) and others indirectly by the calcium receptor protein, calmodulin. These pathways are integral to muscle contraction, neuroendocrine secretion, cell metabolism and growth. The total body calcium in a human adult is approximately 1 kg, 99% of which is contained in the skeleton. Approximately 1% of skeletal calcium is freely exchangeable with calcium in the extracellular fluid (ECF). Calcium ions diffuse freely throughout the extracellular space, where their concentration is approximately 1.2 mmol/L. The plasma ionized calcium concentration is the same, but the plasma total calcium is approximately two-fold higher because of protein binding of calcium. Calcium also forms complexes with phosphate, citrate and bicarbonate in plasma and interstitial fluid (Table 6.1). It is the ionized calcium concentration that is physiologically important and closely regulated. TABLE 6.1 Distribution of plasma calcium Calcium is principally an extracellular ion; cytosolic concentrations are of the order of 10− 4 to 10− 3 mmol/L. The low intracellular calcium concentration is necessary for calcium to function as an intracellular messenger. It is maintained by calcium pumps and exchangers on cell membranes. The endoplasmic reticulum and the mitochondria also have the capacity to remove calcium from the cytosol. Three principal organs are involved in the body’s handling of calcium: the gastrointestinal tract, bone and the kidneys (Fig. 6.1). Intestinal absorption of calcium is mediated by two mechanisms. One is an active transcellular process, regulated by calcitriol (1,25-dihydroxyvitamin D; 1,25(OH)2D) in the duodenum. This involves uptake of calcium into the enterocyte by TRPV6 (membrane calcium channel transient receptor potential cation channel, subfamily V, member 6), followed by intracellular binding of calcium to the calcium-binding protein CaBP-9k, then energy-dependent transport of the calcium across the basolateral membrane via PMCA1b, an alternatively spliced transcript of plasma membrane Ca2 +-ATPase. 1,25-Dihydroxyvitamin D increases gene expression of TRPV6 and CaBP-9k, thus increasing calcium absorption. Calcium is also absorbed passively throughout the small intestine and possibly in the colon. Because the duodenum is relatively short compared to the rest of the small bowel, it may account for less than half of the calcium absorbed at normal dietary intakes, despite its greater absorptive capacity per unit length. At low calcium intakes, active absorption predominates, but this is saturable, and at higher intakes, the change in net absorbed calcium in relation to any change in intake is relatively small and mediated by passive paracellular absorption. The gut not only absorbs calcium but also secretes it in the digestive juices. The unabsorbed portion of this secreted calcium (approximately 2–3 mmol/24 h) is known as the endogenous faecal calcium. Thus at very low intakes, the absorbed dietary calcium will be less than that lost from secretion in digestive juice and the net calcium absorption will be negative. Net calcium absorption is positive in healthy adults when their daily calcium intake is greater than 5 mmol. The absorption of calcium is influenced by other dietary constituents. The presence of anions such as phosphate, oxalate (found in some fruit and vegetables) and phytate (found in some unrefined cereals) diminishes calcium solubility and thus net absorption. Gastrointestinal absorption of calcium tends to decline with age, but is increased during pregnancy and lactation. The principal regulator of intestinal absorption is 1,25(OH)2D, and so either deficiency or excess of this hormone is associated with parallel changes in calcium absorption. The average calcium intake of healthy adults consuming a Western diet is about 20 mmol/day, of which 20–40% is absorbed. The principal source of calcium in the Western diet is dairy products. There are significant amounts of calcium in some green vegetables such as spinach, though the bioavailability is lower. The ionized and complexed fractions of plasma calcium are filtered at the glomeruli, amounting to approximately 250 mmol/24 h. Of this, ~ 98% is reabsorbed, mostly in the proximal tubules. Calcium reabsorption in the proximal tubules is through a paracellular, therefore passive, process, which is closely linked with that of sodium and does not appear to be hormonally regulated. The fine-tuning of calcium excretion occurs in the distal parts of the nephrons where ~ 15% of the filtered load is reabsorbed. This section consists of the distal convoluted tubules, the connecting tubules and the initial portion of the cortical collecting ducts. In these distal sites, calcium reabsorption is active and occurs against an electrochemical gradient. Active calcium reabsorption is a multistep process with initial passage of Ca2 + across the luminal membrane through the epithelial channel TRPV5 (membrane calcium channel transient receptor potential cation channel, subfamily V, member 5), then cytosolic diffusion bound to vitamin D-sensitive calcium-binding proteins (calbindins), and finally active extrusion across the opposite basolateral membrane by a Na+, Ca2 +-exchanger (NCX1) and/or Ca2 +-ATPase (PMCA1b). This active calcium reabsorption is subject to hormonal regulation – principally by parathyroid hormone (PTH) and extracellular calcium itself, but possibly also by 1,25(OH)2D, calcitonin, oestrogens and androgens. Urine calcium excretion is increased in subjects consuming a high protein diet, because of acid production in the metabolism of sulphur-containing amino acids and sequestration of calcium by sulphate in the urine, resulting in inhibition of calcium reabsorption. Sodium intake can also influence calcium excretion by affecting proximal tubular sodium and calcium reabsorption (increased sodium intake increasing calcium excretion). Other factors that increase renal tubular calcium reabsorption are hypovolaemia, alkalosis and thiazide diuretics, whereas volume expansion, acidosis and loop diuretics (e.g. furosemide) have the opposite effect. In the mature adult, the movement of calcium into bone equals its rate of efflux and bone mass remains constant. The situation is different in both growth and senescence. In spite of this constancy of adult bone mass, there is an active exchange of calcium between bone and the ECF. This can take place as a result of bone remodelling (see Chapter 31), or it can be accomplished by a process of mineral exchange between bone and the ECF, without local changes in bone matrix. Bone remodelling accounts for the changes in bone density that take place with ageing or disease. In the healthy adult, about 5% of the entire skeleton is remodelled in one year. In contrast, radioisotope studies have indicated that 1–2% of total body calcium can be exchanged between the bone and ECF over a period of several days. The precise mechanisms of this exchange and the factors influencing it are not known, but the quantities of calcium involved suggest that it may be an important part of normal calcium homoeostasis. Calcium losses in sweat are in the order of 0.3 mmol/24 h. Breast milk has a high calcium content (~ 7.5 mmol/L) and a breast-feeding woman can lose 4–8 mmol/24 h in her milk. Plasma calcium concentration is principally controlled by PTH and 1,25(OH)2D. Calcitonin may also be regarded as a calcitropic hormone, although it has no clearly defined physiological function in humans. Many other hormones, growth factors and cytokines can influence bone metabolism. Parathyroid hormone is an 84-amino acid, single-chain polypeptide secreted by the parathyroid glands. The gene is on chromosome 11 and consists of three exons encoding a peptide of 115 amino acids (pre-pro-PTH), which is cleaved to produce pro-PTH (90 amino acids) and subsequently the mature peptide (84 amino acids), before secretion. Most of the biological activity of PTH appears to reside in the 34 amino acids at the N-terminal end of the peptide. The principal regulator of PTH secretion is the ECF ionized calcium concentration: low concentrations stimulate secretion and high concentrations inhibit it. This regulation is mediated by the calcium sensing receptor (CaSR), a cell membrane-bound, G-protein-coupled receptor. The relationship between PTH secretion and ionized calcium concentrations is not linear. Marked hysteresis is evident from experiments where ionized calcium concentrations are raised or lowered. The PTH concentration at any given concentration of plasma ionized calcium is lower when the ionized calcium concentration is rising than it is when ionized calcium is falling (Fig. 6.2). Plasma PTH concentrations exhibit a diurnal variation. They are stable during the afternoon and evening, but then rise by about 50% to peak at around 02.00 h and subsequently fall to values approximately 50% below afternoon values around 09.00 h. The zenith to nadir difference (in healthy men) is ~ 2.3 pmol/L, using an intact PTH assay. Many other factors have been shown to influence PTH secretion, including 1,25(OH)2D, adrenergic agonists, prostaglandins and magnesium, but whether any of these are physiologically important is debatable. Severe magnesium deficiency can produce a state of reversible hypoparathyroidism. Parathyroid hormone binds to cell surface receptors in its target tissues. The PTH receptor has seven transmembrane domains, an extensive extracellular domain involved in hormone binding and an intracellular domain regulating intracellular signalling. In its two principal target tissues, bone and kidney, this results in activation of both adenylate cyclase (with production of cyclic adenosine monophosphate, cAMP) and phospholipase C (with production of inositol trisphosphate) and subsequent mobilization of intracellular calcium. In bone, PTH receptors are present on cells of osteoblastic lineage. The osteoblasts in turn regulate osteoclast maturation through production of RANKL (receptor activator of nuclear factor κ-B ligand), which binds to the receptor, RANK, on pre-osteoclasts, leading to their maturation to osteoclasts. This process is modulated by osteoblast production of osteoprotegerin (OPG), which can also bind RANK-ligand, thus reducing its binding to RANK and so the stimulation of osteoclastogenesis. Parathyroid hormone has three principal actions on the kidneys. It reduces proximal tubular reabsorption of phosphate, increases distal tubular reabsorption of calcium and increases the activity of the 25-hydroxyvitamin D 1α-hydroxylase enzyme in proximal tubular cells. It also decreases proximal reabsorption of bicarbonate, leading to a mild hyperchloraemic acidosis in states of PTH excess. The development of assays for PTH has greatly simplified the investigation of patients with abnormal plasma calcium concentrations, but these assays do have limitations. The parathyroid gland secretes both the intact 84-amino acid peptide and inactive PTH fragments. Hepatic and renal metabolism of intact PTH results in the appearance of mid-region and carboxy-terminal peptides in the circulation. In vivo, the principal biologically active peptide is intact parathyroid hormone, which is present at a low concentration (1–5 pmol/L) and has a short half-life (2–4 min). However, mid-molecule and carboxy-terminal fragments have significantly longer half-lives and accumulate in concentrations 5–20 times greater than that of the intact peptide. These inactive fragments are cleared by the kidneys and their concentrations increase substantially in the presence of renal insufficiency. Although these fragments are not biologically active, they may be immunoreactive, if they cross-react with the particular antibodies used in the immunoassay. Assays that detect inactive fragments, therefore, report higher PTH concentrations than those that detect the intact molecule. In recent years, two-site and immunoassays of ‘intact’ PTH, which purportedly measure only the biologically active intact peptide, have been widely used. However, there is evidence that these first-generation assays for intact PTH also detect non-(1–84)-PTH peptides, in particular PTH 7–84, which may have different biological actions from the full-length peptide. Assays based on antibodies directed at the first four amino acids of PTH (second-generation intact PTH assays) reveal that non-(1–84)-PTH peptides account for about 15% of circulating immunoreactive PTH (as detected by first-generation assays) in healthy patients, and at least 30% in patients with either primary or secondary hyperparathyroidism. There is also evidence that some non-intact peptides might be biologically active, possibly antagonizing the actions of the intact hormone. Whether assays based on antibodies directed at the extreme N-terminus of PTH offer important clinical advantage in the management of either primary hyperparathyroidism or renal bone disease over the widely available first-generation intact PTH immunoassays remains uncertain. Both mid-molecule and intact PTH assays show an increase in reference values with age, though this is more marked for the mid-molecule assays because of increased fragment accumulation secondary to the age-related decline in renal function. Because of the effect of renal function on measurements using mid-molecule and carboxy-terminal PTH assays, their interpretation must always take into account the patient’s estimated GFR. Owing to the heterogeneity of PTH immunoassays currently in use, it is important that laboratories use assay-specific data for reference ranges and the ranges of values to be expected in hyperparathyroidism, hypercalcaemia of malignancy, hypoparathyroidism and renal disease. Failure to do so may result in inappropriate management of patients with renal disease. The action of PTH on the kidney causes cAMP to appear in the urine. ‘Nephrogenous’ cAMP has been used in the past as a surrogate measure of PTH. Its current use is limited to the evaluation of PTH resistance (see Appendix 6.3, below). Increases in plasma concentrations of PTH are seen in a variety of circumstances. It is useful clinically to distinguish conditions in which the increased secretion of PTH is a normal physiological response to hypocalcaemia (secondary hyperparathyroidism) from those in which hypersecretion of PTH is the principal abnormality (primary hyperparathyroidism). If secondary hyperparathyroidism persists over long periods then the parathyroids can become hyperplastic. A gradual rise in plasma calcium accompanies this change and overt hypercalcaemia can develop. This is known as tertiary hyperparathyroidism, and arises chiefly in long-term dialysis patients, but it has also been described in patients with malabsorption syndromes. This classification is unsatisfactory in some respects. For example, the kidney transplant recipient with hypercalcaemia due to hyperparathyroidism that developed while on dialysis is difficult to categorize. An alternative classification, which has the attraction of being more closely related to treatment stratagems, is to describe hyperparathyroidism in relation to the prevailing plasma calcium concentration. Thus, hyperparathyroidism can be described as hypocalcaemic, normocalcaemic or hypercalcaemic. Vitamins are defined as essential organic compounds that the body cannot synthesize, and are therefore a vital component of the diet. The term ‘vitamin D’ is a misnomer as the majority of vitamin D is synthesized in the skin by the action of ultraviolet light on 7-dehydrocholesterol (Fig. 6.3). This produces cholecalciferol (vitamin D3). Vitamin D is present in a variety of foodstuffs. It can be added to food in the form of ergocalciferol (vitamin D2 – an additive to margarine and some breakfast cereals and, in the USA, to milk) or may occur naturally in animal products in the form of cholecalciferol. Fish oils are the richest dietary source of cholecalciferol. In most respects, these two forms of vitamin D have similar metabolism and action. The calciferols are fat soluble and large quantities can be stored in adipose tissue. They are virtually without biological activity unless they have been hydroxylated. Hydroxylation by the enzyme vitamin D 25-hydroxylase (CYP2R1), takes place in the liver producing 25-hydroxyvitamin D (25(OH)D, calcidiol), which is the principal circulating metabolite and the prohormone for 1,25(OH)2D (calcitriol). The dependence on dermal synthesis for provision of vitamin D is well illustrated by the marked variation in plasma 25(OH)D concentrations through the year in non-tropical areas of the world, where values are significantly higher in summer and autumn than in winter and spring. The vitamin D metabolites circulate in plasma bound to an α-globulin, vitamin D-binding protein and, to a lesser extent, to albumin (Table 6.2). TABLE 6.2 Circulating concentrations of vitamin D metabolites a Higher in summer/autumn than winter/spring; b higher in pregnancy and during puberty. c Vitamin D binding protein. Interconversions: 10 nmol ≡ 4 μg ≡ 160 international units. Most of the circulating 25(OH)D is metabolized in the liver via intermediates such as 23,25(OH)2D and 25,26(OH)2D into inactive metabolites that are excreted into the bile. A small proportion of circulating 25(OH)D undergoes further hydroxylation in the cells of the proximal renal tubules to produce either 1,25(OH)2D or 24,25(OH)2D (secalciferol). The principal active metabolite of vitamin D is 1,25(OH)2D. Although circulating concentrations of 24,25(OH)2D are normally ten times greater than those of 1,25(OH)2D, it has no definite physiological role. In contrast to hepatic 25-hydroxylation, renal 1α-hydroxylation is closely regulated, being enhanced by low circulating concentrations of phosphate and 1,25(OH)2D, and by high concentrations of PTH, and suppressed by high concentrations of the bone-derived hormone fibroblast growth factor 23 (FGF23). When the plasma concentration of phosphate is high and PTH low, 1α-hydroxylase activity (enzyme CYP27B1) decreases and that of 24-hydroxylase (enzyme CYP24B) becomes greater. Other factors may also influence renal hydroxylation of vitamin D. In vitro, low calcium can stimulate 1α-hydroxylation independently of PTH, although whether this is significant in vivo is not known. Direct or indirect effects of growth hormone, prolactin, oestrogens and calcitonin have also been described. 1,25-Dihydroxyvitamin D is inactivated by the formation of more polar metabolites, which are excreted primarily into bile. Over 96% of all vitamin D is ultimately eliminated through the bile into the faeces. Since this excretion is largely in the form of inactive metabolites, it is doubtful if there is any enterohepatic recycling of physiological importance. Although the kidneys are undoubtedly the major source of production of 1,25(OH)2D, there is evidence from anephric haemodialysis patients that small quantities can be synthesized extrarenally. In pregnancy, 1,25(OH)2D is synthesized in the placenta, and in a variety of granulomatous diseases (sarcoidosis, tuberculosis, berylliosis) the macrophages of the granulomata can also 1α-hydroxylate vitamin D. In these circumstances, the synthesis is substrate dependent, meaning that the more 25(OH)D that is available, the more 1,25(OH)2D is synthesized. Vitamin D metabolites act via a cytosolic receptor that is translocated to the nucleus where it regulates gene expression. The vitamin D receptor is similar in structure to the steroid and thyroid hormone receptors, having an amino-terminal domain, a central DNA-binding domain with ‘zinc fingers’ and a carboxy-terminal hormone-binding domain. 1,25-Dihydroxyvitamin D has an affinity for the vitamin D receptor that is approximately 1000 times greater than that of 25(OH)D, which in turn is an order of magnitude more potent than 24,25(OH)2D. However, the circulating concentration of 25(OH)D is substantially greater than that of 1,25(OH)2D, so it is possible that this metabolite has some biologically significant effect. The principal site of action of 1,25(OH)2D is the intestine, where it stimulates the production of the calcium channels TRPV6 and TRPV5, and the calcium-binding protein CaBP-9k. In bone, it is a potent osteolytic factor in vitro, acting through osteoblast RANK-ligand production to bring about fusion of osteoclast precursors to form mature osteoclasts. The physiological importance of its bone resorbing effect is uncertain. 1,25-Dihydroxyvitamin D also has direct effects on the liver and kidney. In the liver, it enhances the production of inactive 25(OH)D derived metabolites that are excreted in the bile, and in the kidney it stimulates the 24,25-hydroxylase – both these mechanisms protect against chronically high 1,25(OH)2D concentrations. Several cells and tissues other than those involved in calcium and phosphorus metabolism, including macrophages, activated T-lymphocytes, the placenta and keratinocytes, express vitamin D receptors and/or the 1α-hydroxylase enzyme, suggesting that their functions might be regulated by components of the vitamin D system. Many malignant cells also express the vitamin D receptor. There is evidence that 1,25(OH)2D decreases the growth of several cancer cell lines, and slows or prevents the growth and/or development of cancers and autoimmune diseases in rodents. Genetic ablation of vitamin D signaling, by deleting the genes encoding either the vitamin D receptor or 1α-hydroxylase, leads to renin-dependent hypertension and an increased propensity to tumour development. The consequences of complete absence of vitamin D signaling in rodents, however, are likely to be very different from minor decreases in 25(OH)D in humans. At present, there is no robust clinical trial evidence that vitamin D supplements or bioactive vitamin D analogues favourably influence important outcomes such as cancer, hypertension or autoimmune diseases. Accordingly, vitamin D supplements are not indicated in the management of non-skeletal diseases. A variety of 1,25(OH)2D analogues have been synthesized. 1α-Hydroxyvitamin D3 (alfacalcidol) is rapidly metabolized by the liver into 1,25(OH)2D and, although having roughly half the potency of 1,25(OH)2D on a weight basis, is similarly useful in conditions where the 1α-hydroxylation step is impaired. A similar preparation, 1α-hydroxyvitamin D2 (doxercalciferol) is also marketed. Other analogues such as 26,27-hexafluorocalcitriol are more potent calcaemic agents. A number of analogues with attenuated calcaemic properties have also been developed. Examples include calcipotriol (used in the treatment of psoriasis), paricalcitol (used in some countries in the treatment of hyperparathyroidism in chronic renal failure) and eldecalcitol (which has been investigated in osteoporosis). The divergent effects of these analogues on cell differentiation, PTH secretion and calcium homoeostasis probably reflect pharmacokinetic differences rather than distinct 1,25(OH)2D-sensitive pathways, since there is no evidence that they act through different receptors. Measurements of 25(OH)D and 1,25(OH)2D can be of value in clinical practice, though the indications for 1,25(OH)2D measurement are relatively few (e.g. obscure causes of hypercalcaemia). 25-Hydroxyvitamin D is the best measure of vitamin D status. While its synthesis is dependent upon 25-hydroxylation in the liver, even advanced liver disease does not seem to be a limiting factor in its production. Free concentrations are normal in most patients with cirrhosis, though total concentrations may be low because of associated hypoproteinaemia. 1,25-Dihydroxyvitamin D is present in concentrations only 1000th of those of its precursor (see Table 6.2). Its production is closely regulated and so is not tied to concentrations of its precursor, except in patients with severe vitamin D deficiency or in granulomatous disease. Renal disease has a major impact on its circulating concentrations, as do states of parathyroid over- or underactivity. A variety of assays have been used in the past, and a number have been withdrawn from the market because of unsatisfactory performance characteristics. Cross-calibration of available assays has not always been within acceptable limits. The reference method uses liquid chromatography with tandem mass spectrometry (MS/MS), but as this is an expensive assay to run, many laboratories use immunoassays based on automated platforms. The acceptability of this compromise depends on the performance characteristics and calibration of the particular assay platform. Some of the commercially available assays have limited ability to measure 25(OH)D2, which may be important when supplementation has been provided in the form of vitamin D2. It is evident that significant variation can occur in measurement of vitamin D metabolites, as a result both of differences between the available assays and in the implementation within individual laboratories. Calcitonin is a 32-amino acid peptide produced in humans in the parafollicular cells (C cells) of the thyroid gland. It contains a disulphide bridge and a proline-amide group at the C-terminus. Both of these and the full amino acid sequence are necessary for biological activity. It is metabolized in the kidneys and has a plasma half-life of approximately 5 min. Secretion of calcitonin is stimulated by an increase in plasma calcium concentration and it is also released in response to a number of gut hormones (e.g. gastrin, glucagon, secretin, cholecystokinin-pancreozymin). It reduces plasma calcium concentration by a direct effect on osteoclasts, which have a cell surface calcitonin receptor linked to adenylate cyclase. It causes contraction of osteoclasts and an acute reduction in osteoclastic bone resorption. It also acts on the kidneys, where it decreases renal tubular reabsorption of calcium and phosphate. Despite these clearly documented actions, its physiological significance is uncertain because its actions appear to be transient. Furthermore, chronic calcitonin deficiency (as in patients post-thyroidectomy) or excess (as in medullary carcinoma of the thyroid) do not result in significant changes in bone or mineral metabolism. There are a number of different forms of calcitonin in the circulation, many of which are not biologically active. Thus, different immunoassays may detect different molecular species. The measurement of calcitonin does not form a significant part of the clinical evaluation of calcium metabolism, but it is an important tumour marker for medullary carcinoma of the thyroid. Procalcitonin, the 116 amino acid peptide precursor of calcitonin, is produced not only by thyroid C-cells but also by the neuroendocrine cells of the lung and the intestine. The amount of procalcitonin produced by the cells of the lung and the intestine increases in response to proinflammatory stimuli, especially of bacterial origin. Measurement of procalcitonin can be used as a marker of severe sepsis and generally correlates well with the degree of sepsis. A number of hormones influence calcium and bone metabolism in vitro, in normal physiology or in specific pathological circumstances. The oestrogen deficiency that develops at the time of the menopause results in increases in cytokine production which leads to increases in bone resorption. Direct effects of oestrogen on osteoclasts and on osteoblastic production of RANKL and osteoprotegerin are also important. Calcium absorption in the gut and reabsorption in the kidney are both reduced, and plasma calcium concentrations rise. These changes lead to a reduction in bone mass, but are reversible with oestrogen replacement therapy. Similarly, testosterone deficiency in men predisposes to osteoporosis, partly through loss of its direct anabolic effects on muscle and on osteoblasts, and partly through diminished conversion to oestrogen. Glucocorticoids can cause marked osteoporosis. These hormones act at a number of sites and reduce osteoblastic activity, induce apoptosis of osteoblasts and osteocytes, reduce intestinal calcium absorption, increase urinary calcium loss and probably increase bone resorption. In children, glucocorticoid excess causes marked growth retardation. Glucocorticoid deficiency may be associated with hypercalcaemia. Growth hormone accelerates linear growth in children and increases plasma phosphate concentration by increasing renal tubular reabsorption of phosphate. These effects are probably mediated by growth hormone-induced changes in the production of insulin-like growth factor 1 (IGF-1, somatomedin C). There is controversy as to the effect of the growth hormone–somatomedin axis on bone density. Subjects with high total body fat mass have been shown to have higher bone densities. This appears to be mediated by a number of hormonal mechanisms. Insulin is anabolic to bone in vitro, and obesity is usually associated with hyperinsulinaemia. At least two other anabolic peptides, amylin and preptin, are co-secreted with insulin and may contribute to the relationship. Leptin has been shown in vitro to stimulate osteoblast growth directly and to inhibit osteoclast development, so it is also likely to be an important factor. In contrast, when leptin is administered into the third ventricle of the brains of experimental animals, there is a profound reduction in appetite and fat mass, with resultant loss of bone mass. There is some evidence that central leptin administration activates the sympathetic nervous system and that this might also contribute to bone loss. Thyroid hormones are potent stimulators of bone resorption, and thyrotoxicosis can be associated with decreased bone density and hypercalcaemia. Prostaglandins of the E series also stimulate bone resorption in vitro. Parathyroid hormone-related peptide (PTHrP) is a 141-amino acid peptide that is responsible for a large proportion of cases of humoral hypercalcaemia of malignancy. Eight of the 13 amino-terminal amino acids are identical to those found in PTH itself, and it appears to act on the PTH receptor. There is evidence for its production in both the fetal parathyroids and in the lactating breast, raising the possibility that it has a physiological role in these contexts. A carboxy-terminal fragment of PTHrP may act as an inhibitor of osteoclastic bone resorption. Direct measurement of PTHrP by immunoassay is possible, but is rarely necessary in clinical practice, since the malignant disease underlying humoral hypercalcaemia is frequently evident at the time that the elevated plasma calcium is recognized. In addition to the assays of calcitropic hormones discussed above, estimation of calcium concentrations in the circulation and of calcium fluxes in the gut and kidney are of value in assessing patients with disorders of calcium metabolism. As indicated previously, total plasma calcium comprises protein-bound, complexed and ionized fractions. Thus changes in plasma protein concentrations or acid–base status will affect the relationship between ionized and total calcium. Total serum calcium remains the most commonly measured index. It should always be accompanied by a measurement of the serum albumin concentration from which a ‘corrected serum calcium’ can be calculated: Laboratories should derive their own correction formulae based on their own particular calcium and albumin assays, as both the mean normal albumin concentration (assumed here to be 40 g/L) and the amount of calcium bound to each gram of albumin (assumed here to be 0.02 mmol/g) can vary. The corrected calcium is only an approximation, and when there are significant dysproteinaemias or acid–base disturbances, ionized calcium should be measured directly. This measurement is also useful in patients to whom large quantities of citrated blood products have been administered, since these individuals have an increased proportion of plasma calcium complexed to citrate. Ionized calcium is measured using an ion-selective electrode. Specimens must be obtained anaerobically to minimize changes in specimen pH. The pH is also usually measured and results adjusted to a pH of 7.4. There are diurnal variations in both total and ionized plasma calcium concentrations (Fig. 6.4). Significant changes in plasma calcium take place following the ingestion of a calcium-rich meal or of calcium supplements. Posture and the application of tourniquets, both of which can influence plasma protein concentrations, may also influence total calcium estimations. Total plasma calcium rises significantly (by approximately 0.1 mmol/L) at the menopause. This is not routinely measured in clinical practice, but knowledge of it is sometimes of value, e.g. in some patients with kidney stones and hypercalciuria. In the past, it was measured by metabolic balance, which required a period of 7–10 days as an inpatient, during which time subjects were given a diet with a constant calcium intake, and calcium absorption was estimated from precise measurements of faecal and urinary calcium output. Calcium absorption can be measured with a variety of stable or radioisotope techniques in which an oral calcium load with a tracer is administered in the fasting state and absorption efficiency is estimated from the rate of appearance of the isotope in the blood. The size of the oral calcium load accompanying the isotope is important. Low doses reflect better the active duodenal transport, whereas high doses better reflect net intestinal absorption. The increment in urine calcium following the administration of a known calcium load has also been used as an index of calcium absorption (see Appendix 6.1, below). The clinical utility of these tests is limited and they are rarely used nowadays. Calcium can be measured either in a 24 h urine or in urine collected after an overnight fast. In the latter instance, the subject empties the bladder on rising, has a glass of water and then collects the urine sample at any time between 30 min and 2 h later. In both instances, acid (usually 6 M hydrochloric acid) is added either before collection or subsequently, to prevent crystallization of calcium salts. Creatinine is usually measured on both 24 h and fasting urine samples – in the former case to allow assessment of completeness of collection and in the latter as a denominator in relation to which the concentration of calcium (and other urinary constituents) can be expressed. The reference range for 24 h urine calcium is 1–7.5 mmol/24 h in men and 1–6.25 mmol/24 h in women. In the steady state, this reflects the absolute dietary intake of calcium and the net proportion absorbed from the intestine. Urine calcium excretion is usually increased with hypercalcaemia of any cause, and its measurement contributes little to the differential diagnosis of hypercalcaemia, except in the diagnosis of familial hypocalciuric hypercalcaemia. Fasting urine calcium can be expressed as a simple molar ratio to creatinine (reference values 0.10–0.30 in adults). This is considered to be an index of bone resorption, but newer markers of bone resorption are preferred in clinical practice (see Chapter 31). If the calcium: creatinine ratio is multiplied by the plasma creatinine concentration, the resulting product is the calcium excretion per litre of glomerular filtrate (CaE). This is useful in assessing the contribution of abnormal renal tubular handling of calcium to disorders of plasma calcium homoeostasis (see Appendix 6.2, below). Disorders of plasma calcium are often accompanied by abnormalities of bone turnover, so assessment of bone turnover can be useful. These measures are discussed in more detail in Chapter 31. Hypercalcaemia develops when the rate of entry of calcium into the ECF from bone and gut exceeds the capacity of the kidney to excrete it. Diminished excretory capacity can be the result of either increased renal tubular reabsorption (an important component, for example, in the hypercalcaemia of primary hyperparathyroidism and familial hypocalciuric hypercalcaemia) or of a reduction in glomerular filtration rate (in sarcoidosis, for example, hypercalcaemia is more frequently encountered in subjects with impaired renal function). However, hypercalcaemia itself can affect these processes, thus initiating a vicious cycle with worsening hypercalcaemia. By its actions on the kidney, hypercalcaemia leads to polyuria, volume depletion and thus to a reduction in glomerular filtration rate and enhanced proximal tubular reabsorption of sodium, which carries calcium with it. This situation, termed ‘disequilibrium hypercalcaemia’, is a medical emergency. It is important both to recognize this and to differentiate it from ‘equilibrium hypercalcaemia’ (e.g. due to mild primary hyperparathyroidism or familial benign hypercalcaemia), where the plasma calcium remains stable. Whether hypercalcaemia causes symptoms depends both upon the degree of elevation of plasma calcium and the rate at which it has risen. When plasma calcium is < 3.0 mmol/L, a large proportion of patients are asymptomatic. The clinical features of severe hypercalcaemia are summarized in Box 6.1. Box 6.2 lists the most frequently encountered forms of hypercalcaemia. Primary hyperparathyroidism is the most common cause of hypercalcaemia presenting outside hospital. Prior to the advent of large automated analysers, it was regarded as a rare disease, but is now a common incidental finding. Its prevalence is approximately 1 per 1000 and it is most frequently diagnosed in the 6th decade of life, when it is two or three times as common in women as in men. To some extent, this can be explained by the postmenopausal rise in plasma calcium concentration. The normal distribution curve is shifted to the right and, therefore, a higher proportion of postmenopausal women must necessarily have plasma calcium concentrations above the upper limit of normal for younger people. In younger patients, the sex incidences are equal. In 90% of patients, primary hyperparathyroidism is attributable to a single parathyroid adenoma, and most of the remaining cases are attributable to four gland hyperplasia, sometimes as a part of the multiple endocrine neoplasia (MEN) syndrome type 1 (see Chapter 41). This syndrome accounts for the majority of patients with familial hyperparathyroidism. Parathyroid carcinoma, which usually presents with severe hypercalcaemia and very high PTH concentrations, accounts for < 1% of all cases. Adenomatous primary hyperparathyroidism appears to have a clonal origin. Two genetic mechanisms have been elucidated (Table 6.3). The PTH gene is located on the opposite side of the centromere of chromosome 11 from the gene (PRAD1) that encodes cyclin D1, a proto- oncogene that regulates cell growth. In about 20% of sporadic parathyroid tumours, it has been shown that while one chromosome is intact, the other copy has undergone centromeric inversion, a rearrangement that places the PTH regulatory elements under the influence of cyclin D1, allowing deregulated parathyroid gland growth. Germline mutations underlie several of the inherited syndromes that include primary hyperparathyroidism. The mutated gene in multiple endocrine neoplasia 1 (MEN1) is also located on chromosome 11, and encodes a protein, menin, that acts as a tumour suppressor. Rare instances have been reported of kindreds with germline MEN1 mutations in which primary hyperparathyroidism is the only clinical feature. Primary hyperparathyroidism is less frequently (~ 20%) a feature of the MEN 2 syndromes, which result from activating mutations of the gene encoding the RET tyrosine kinase receptor. Germline mutations in the tumour suppressor gene CDC73, which encodes the protein parafibromin, causes the inherited disorder, hyperparathyroidism-jaw tumour syndrome. Somatic mutations in this gene are also present in a high proportion of sporadic parathyroid carcinomas. Primary hyperparathyroidism may present with the symptoms of hypercalcaemia outlined in Box 6.1, but frequently patients are symptom-free. The prevalence of osteoporosis is probably increased in patients with primary hyperparathyroidism, especially at skeletal sites enriched for cortical bone. The classic skeletal manifestations of primary hyperparathyroidism, which include subperiosteal phalangeal resorption, a ‘salt and pepper’ appearance in the skull and bone cysts (‘brown tumours’) of the long bones or jaw (collectively termed ‘osteitis fibrosa cystica’), are features of severe, longstanding disease, and are now uncommon in the developed world. Approximately 15% of patients have renal complications in the form of nephrolithiasis or nephrocalcinosis. Familial hypocalciuric hypercalcaemia (FHH) (also known as benign familial hypercalcaemia) is an autosomal dominant condition in which mild hypercalcaemia with relative hypocalciuria is present throughout life. Patients with this disorder are most frequently heterozygous for inactivating mutations in the calcium sensing receptor (CaSR). The ‘set-point’ for PTH release is raised, and mild PTH-dependent hypercalcaemia results. The CaSR is also expressed in the renal tubules, where it regulates renal tubular calcium reabsorption in a PTH-independent fashion. The inactivating CASR gene mutation promotes inappropriately avid tubular calcium reabsorption in the face of hypercalcaemia, explaining the relative hypocalciuria. Patients with FHH are usually asymptomatic, although non-specific symptoms of lethargy and polydipsia have been observed in some instances. There may be an increased incidence of pancreatitis, but the frequency of nephrolithiasis and peptic ulcer disease is the same as in the general population. In most cases, the condition follows a benign course and the greatest danger to patients is being misdiagnosed as having adenomatous primary hyperparathyroidism and thus having inappropriate parathyroidectomy. Familial hypocalciuric hypercalcaemia accounts for about 2% of asymptomatic hypercalcaemia. Neonatal severe primary hyperparathyroidism occurs in newborns who have inherited two inactivating CASR mutations – one from each parent. It presents as severe, life-threatening hypercalcaemia. Urgent total parathyroidectomy is indicated. Malignancy can result in hypercalcaemia by four general mechanisms: •
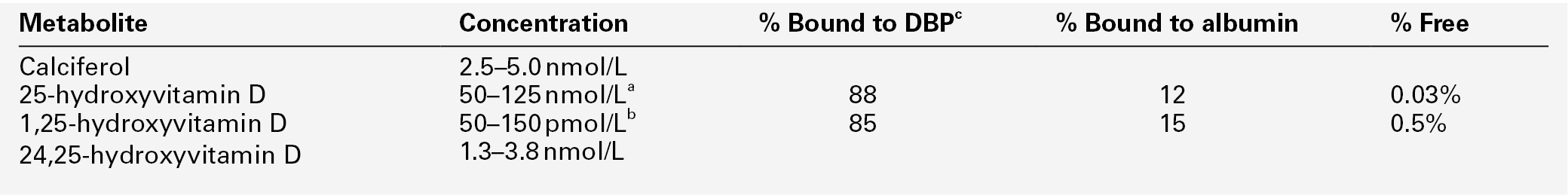
Calcium, phosphate and magnesium
CALCIUM METABOLISM
Biological role of calcium
Distribution of calcium
Fraction
Total (%)
Ultrafilterable
53
Ionized
47
Complexed
6
Protein bound
47
Albumin
37
Globulin
10
Total (2.2–2.6 mmol/L)
100
Calcium fluxes
Gastrointestinal tract
Kidneys
Bone
Regulation of calcium metabolism
Parathyroid hormone
Measurement of circulating parathyroid hormone
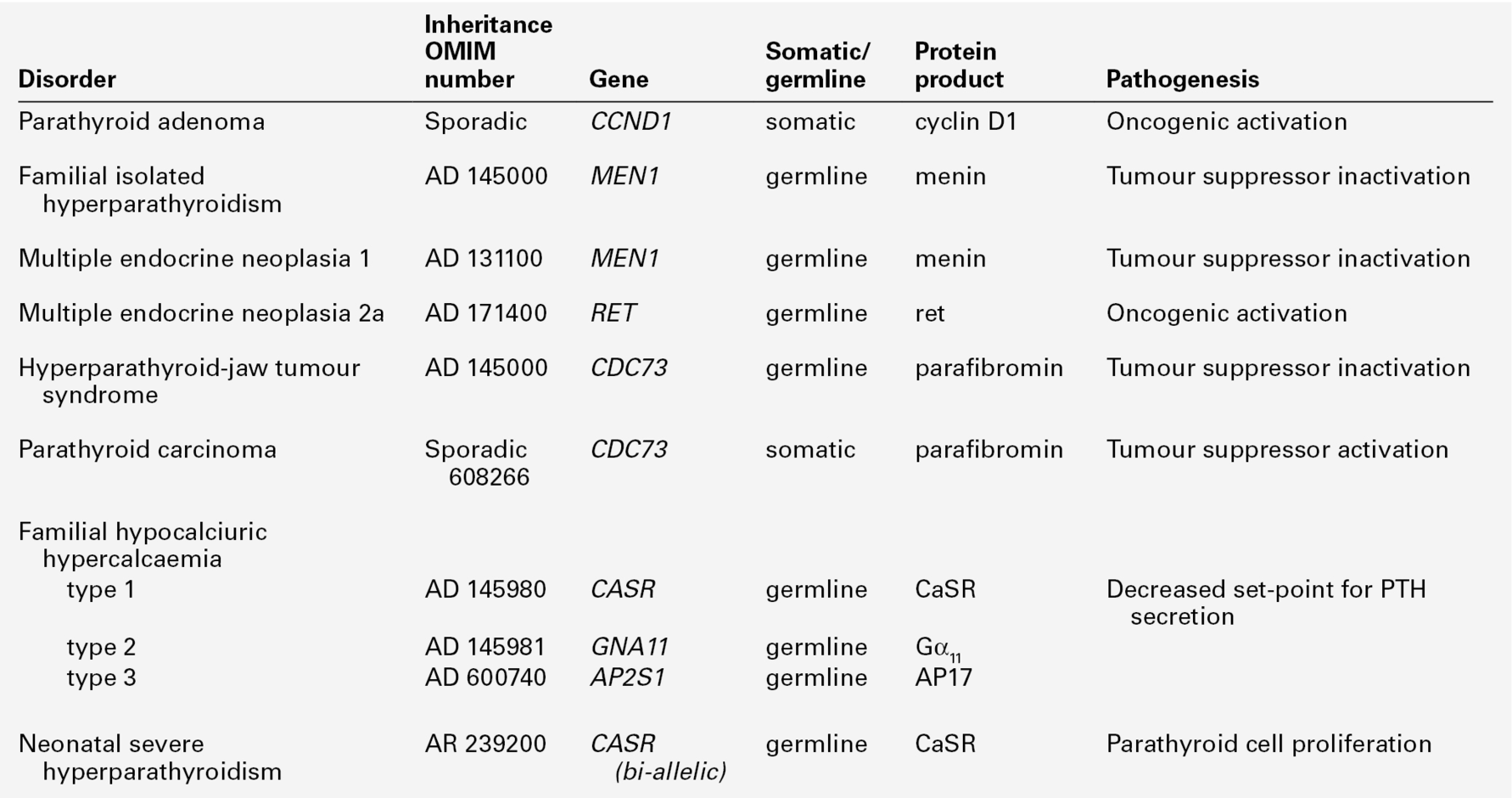
Classification of hyperparathyroidism
Vitamin D
Synthesis and metabolism
Actions
Synthetic vitamin D analogues
Measurement of vitamin D metabolites
Calcitonin
Procalcitonin
Other hormones
Biochemical assessment of calcium metabolism
Plasma calcium

Intestinal calcium absorption
Urinary calcium
Indices of bone turnover
Hypercalcaemia
Causes of hypercalcaemia
Primary hyperparathyroidism
Familial hypocalciuric hypercalcaemia
Hypercalcaemia of malignancy
![]()
Stay updated, free articles. Join our Telegram channel
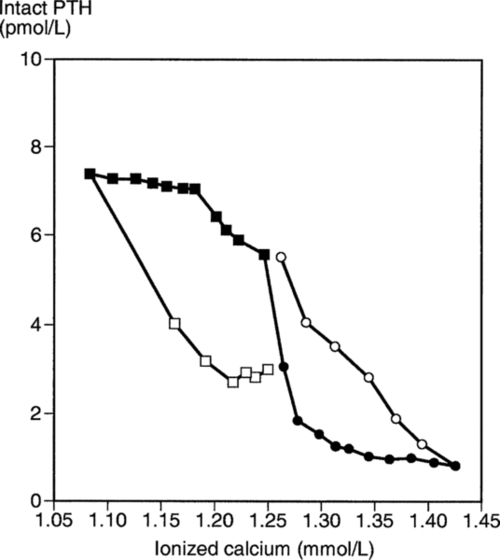
 ), and during recovery from hypocalcaemia (
), and during recovery from hypocalcaemia ( ) and hypercalcaemia (
) and hypercalcaemia ( ). For any given ionized calcium concentration, the PTH concentration is lower when the ionized calcium is rising than when it is falling.
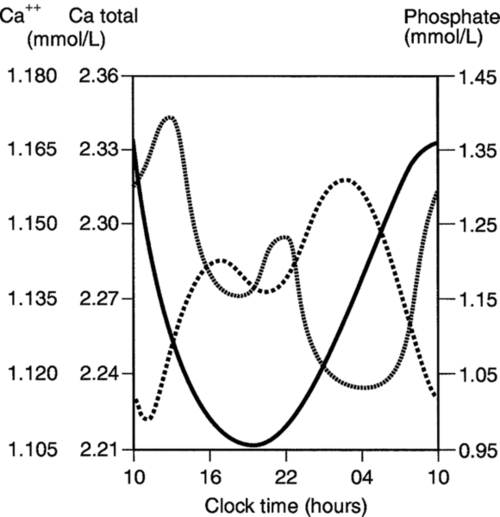
). For any given ionized calcium concentration, the PTH concentration is lower when the ionized calcium is rising than when it is falling. 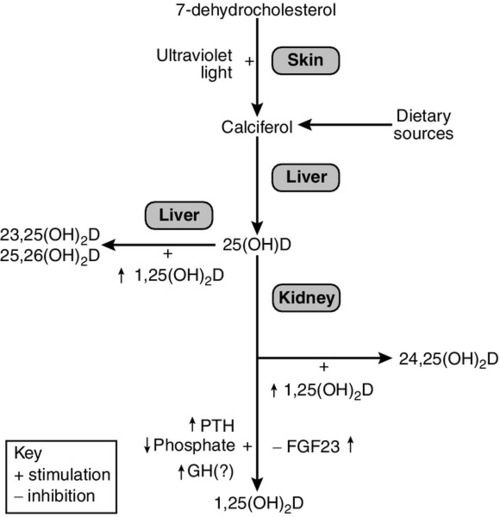

Full access? Get Clinical Tree


