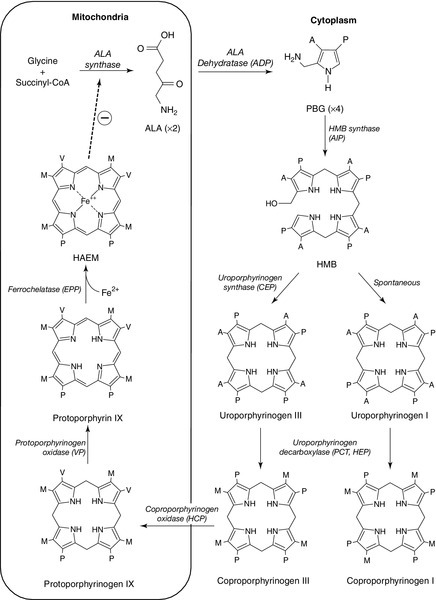
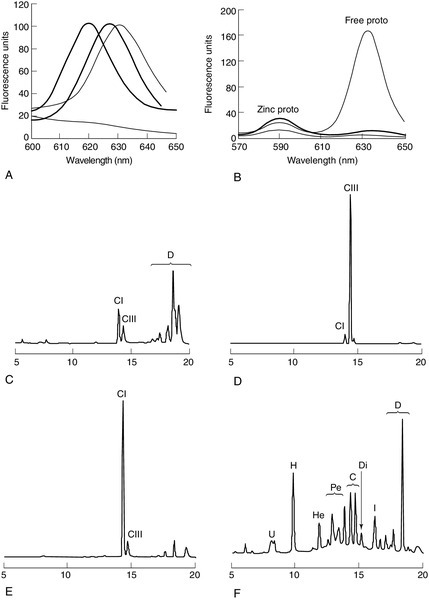
CHAPTER 28 Michael N. Badminton; George H. Elder CHAPTER OUTLINE Biochemistry of haem synthesis Molecular genetics of the porphyrias PORPHYRIAS PRESENTING WITH ACUTE ATTACKS The autosomal dominant acute porphyrias Erythropoietic protoporphyria and X-linked dominant protoporphyria SECONDARY DISORDERS OF PORPHYRIN METABOLISM The porphyrias are a group of eight metabolic disorders that result from inherited or acquired functional abnormalities of enzymes of the haem biosynthetic pathway (Table 28.1 and see Fig. 28.1, below). No disease has yet been associated with defects in the 5-aminolaevulinic acid (ALA) synthase-1 (ALAS1) gene that encodes the ubiquitous isoform of ALAS, the first enzyme of the pathway. However, gain of function mutations in the erythroid-specific ALAS (ALAS2) gene cause X-linked dominant protoporphyria (XLDPP), whereas loss of function causes X-linked sideroblastic anaemia. TABLE 28.1 Overview of the porphyrias indicating inheritance, prevalence and main clinical presentation AR, autosomal recessive; AD, autosomal dominant. a Also known as PBG deaminase. b Enzyme activities are half normal. FIGURE 28.1 The haem biosynthetic pathway. The side chains are denoted by: A, acetic acid; M, methyl; P, propionic acid; V, vinyl. For other abbreviations, see Box 28.1. The principal clinical features of the porphyrias are neurovisceral or cutaneous, or both. This chapter approaches the subject from a practical perspective, describing how and why patients present, how they are diagnosed and how patients and their families should be managed. A list of frequently used abbreviations is provided in Box 28.1. Haem is essential for life and is synthesized in all cells, although the major sources are bone marrow (80%) and liver (15%). Haemoproteins include haemoglobin and myoglobin, which are the most abundant; the mitochondrial respiratory cytochromes; enzymes such as catalase and tryptophan pyrrolase, and the cytochrome P450 enzymes that are components of many essential metabolic processes, including the metabolism of xenobiotics. The haem synthetic pathway comprises eight steps, each catalysed by a specific enzyme, of which the first and last three are mitochondrial and the remainder cytosolic (Fig. 28.1). The first enzyme, ALAS, catalyses the condensation of succinyl-CoA and glycine into 5-aminolaevulinic acid and is the rate-controlling step in all cells. Regulation of ALAS1 in the liver and other non-erythroid tissues is via inhibition by haem, the end product of the pathway, a characteristic that underlies both the pathogenesis and treatment of acute attacks of porphyria. In erythroid cells, regulation of haem synthesis is iron dependent. The second step is the synthesis of porphobilinogen (PBG), a watersoluble, colourless monopyrrole, catalysed by ALA dehydratase. The third step is the polymerization of four molecules of PBG to form the colourless linear tetrapyrrole, 1-hydroxymethylbilane (HMB), by the enzyme hydroxymethylbilane synthase (HMBS, also known as PBG deaminase). This linear molecule is cyclized into the tetrapyrrole ring structure, uroporphyrinogen III, by the enzyme uroporphyrinogen III synthase. The porphyrinogens are colourless, non-fluorescent, unstable compounds, which rapidly oxidize to their red-purple porphyrin equivalents. Porphyrins absorb light, are fluorescent and therefore photosensitizing. These properties also make them relatively straightforward to measure in the clinical biochemistry laboratory. Uroporphyrinogen III, which is hydrophilic by virtue of its eight carboxy groups, is converted to coporphyrinogen III by uroporphyrinogen decarboxylase, which catalyses the sequential removal of four carboxyl residues. Two further carboxyl groups are removed in an oxygen-dependent dehydrogenation-decarboxylation reaction catalysed by the enzyme coproporphyrinogen oxidase (CPOX) to form protoporphyrinogen, which is then oxidized to protoporphyrin IX by protoporphyrinogen oxidase (PPOX). Progressive decarboxylation makes these precursors and the corresponding porphyrins increasingly hydrophobic, which determines their routes of excretion (see Fig. 28.2). The final step in the pathway is insertion of ferrous iron (Fe2 +) into protoporphyrin to form haem, catalysed by ferrochelatase (FECH). In the absence of iron, other divalent cations, such as zinc, may be inserted. Although only the III isomer can progress through the pathway to form protoporphyrin IX and haem, HMB may spontaneously cyclize into the uroporphyrinogen I isomer. This forms a substrate for uroporphyrinogen decarboxylase (UROD) and may be converted to coproporphyrinogen I, but is not metabolized further. Adult reference ranges for porphyrins and their precursors are shown in Table 28.2. FIGURE 28.2 Examples of key laboratory findings that allow the porphyrias to be distinguished biochemically. (A) Plasma porphyrin fluorescence scans showing the three distinct emission peaks at 620 nm (AIP, CEP, PCT, HCP), 626 nm (VP) and 631 nm (EPP). A negative plasma scan (lower line) is also shown. (B) Fluorescence emission scan of whole blood allows zinc chelated protoporphyrin (proto) (increased in anaemia and lead poisoning) to be distinguished from free protoporphyrin, which is increased in EPP. Markedly increased zinc chelated and free protoporphyrins are characteristic for XLDPP. The lower line indicates a normal scan. High performance liquid chromatography (HPLC) analysis of faecal porphyrins allows three porphyrias with a plasma fluorescence emission peak at 620 nm to be distinguished (faecal porphyrin excretion is normal in AIP). (C) Normal faecal porphyrin HPLC trace indicating predominantly coproporphyrin (CI) isomer and dicarboxylic (D) porphyrins (protoporphyrin, pemptoporphyrin, deuteroporphyrin). (D) Faecal porphyrin pattern found in HCP indicating increased coproporphyrin, which is almost entirely the III isomer. (E) Faecal porphyrin trace seen in CEP indicating increased coproporphyrin isomer I. (F) Faecal porphyrin pattern seen in PCT patients indicating the increased excretion of partially decarboxylated intermediates; heptacarboxylic (H), hexacarboxylic (He) and pentacarboxylic (Pe) porphyrins as well as isocoproporphyrin (I) and dehydroisocoproporphyrin (Di), which are pathognomonic for PCT. U, uroporphyrin. Clinically manifest porphyria is always associated with detectable overproduction of haem precursors. Each enzyme deficiency, and the increase in activity in XLDPP, gives rise to a specific pattern of overproduction, which defines the corresponding disease (Table 28.3). Three clinical manifestations may occur: acute neurovisceral attacks, skin lesions or both. Acute attacks of porphyria are always accompanied by overproduction of ALA and, in all but ALA dehydratase deficiency porphyria (ADP), of PBG. Porphyrias causing skin lesions are characterized by overproduction of porphyrins. Four of the eight porphyrias can present with acute neurovisceral attacks: the very rare autosomal recessive ADP and the three autosomal dominant acute porphyrias, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP) and variegate porphyria (VP). Hereditary coproporphyria and VP can present with skin photosensitivity or acute attacks, or both. In the other four porphyrias, two types of photosensitization may occur: accumulation of hydrophobic free protoporphyrin in erythropoietic protoporphyria (EPP) and XLDPP is associated with acute photosensitivity, while accumulation of the more water-soluble porphyrins in porphyria cutanea tarda (PCT) and congenital erythropoietic porphyria (CEP) leads to fragile skin and bullae. TABLE 28.3 The main biochemical findings in symptomatic porphyria Porphyrin analyses on random urine, faeces and EDTA-preserved blood allow the individual porphyrias to be distinguished. Individual urine and faecal porphyrins are separated and measured by high-performance liquid chromatography with fluorescence detection. Note that EPP cannot be diagnosed by urine porphyrin analysis alone. a Emission maximum of plasma fluorescence peak (for examples, see Fig. 28.2). All the porphyrias, apart from the sporadic form of PCT, are single gene disorders that are inherited in autosomal dominant, recessive or X-linked patterns (see Table 28.1). Characteristics and chromosomal locations of individual genes are shown in Table 28.4. The HMBS and UROS genes are alternatively spliced to produce erythroid and ubiquitous isoforms. Disease-specific mutations that abolish or markedly decrease enzyme activity have now been identified in the genes for all the autosomal dominant porphyrias (Human Gene Mutation Database: www.hgmd.org). In most countries, mutational analysis has revealed extensive allelic heterogeneity, with large numbers of mutations identified in each gene, most of which are present in only one or a few families. The main exceptions are the W198X HMBS mutation in Sweden and the R59W PPOX mutation in South Africa. In both countries, these mutations have been multiplied by founder effects and account for the high prevalence of AIP in Sweden and of VP among individuals of Afrikaans ancestry in South Africa. The proportion of different types of mutation varies little between the diseases, with missense, nonsense, splice site and frameshift mutations contributing to the overall heterogeneity. Large deletions appear to be uncommon. Perhaps not surprisingly in view of the large number of different mutations, no clinically useful genotype-phenotype correlation has yet been identified in any autosomal dominant porphyria. With current methods of analysis, the sensitivity of mutation identification is over 95% in AIP and VP. Incomplete penetrance is an important feature of all the autosomal dominant porphyrias; only a proportion of those who inherit a disease-specific mutation develop the disease. In this chapter, the terms latent or presymptomatic will be used to describe individuals who have inherited a porphyria gene but at the time of investigation have not had symptoms. Estimates of penetrance for the autosomal dominant acute porphyrias derived from family studies range from 10% to 40% and are influenced by age and rigour of phenotype definition. In the UK, about 80% of adults identified by family studies as carrying a gene for an acute porphyria never have an acute attack severe enough to require hospitalization. This figure is consistent with the observation that about 30% of patients with AIP present without a family history of the disease, yet family studies almost always reveal latent porphyria in their families, de novo mutation being uncommon. Studies of blood donors suggest that the gene for AIP may be present in about 0.06% of the general population. The low clinical penetrance of the acute porphyrias probably reflects a combination of environmental (see below) and genetic influences. The latter have not been identified but are likely to be at loci distant from the disease gene. Factors that determine clinical penetrance in familial PCT are described on page 544. Clinical penetrance of the autosomal recessive porphyrias, ADP, CEP and EPP is close to 100%, although age at first presentation may be variable. They also show extensive allelic heterogeneity, with most patients whose parents are not consanguineous being compound heterozygotes. Other features of their genetics, and those of XLDPP, are discussed later in this chapter. AIP, HCP and VP are autosomal dominant conditions that put the patient at risk of acute neurovisceral attacks, which may be life-threatening. In VP and HCP, skin lesions, indistinguishable from those of PCT, may occur either independently or in combination with acute attacks. In the UK, acute attacks of porphyria are estimated to affect about nine per million of the population, most of whom have AIP. Symptomatic VP is approximately half as common as AIP. HCP is the rarest of these acute porphyrias. Acute neurovisceral attacks occur when hepatic haem synthesis is induced in the presence of deficient HMBS activity, resulting in an accumulation of the haem precursors ALA and PBG. In AIP, this is a primary deficiency, but in VP and HCP, it has been proposed that the deficiency is secondary to inhibition of HMBS by other metabolites in the haem pathway. The exact cause of neuronal damage has not been fully established, but current evidence from liver transplantation suggests that it is due to a hepatic neurotoxin, probably ALA. The neurological lesions comprise axonal degeneration and patchy demyelination of peripheral neurons with chromatolysis of anterior horn cells, brain stem nuclei and ganglia of the autonomic nervous system. There may also be diffuse neuronal loss and gliosis of the CNS. The result is damage to autonomic, motor and CNS neurons, giving rise to the characteristic clinical presentation, described below. Acute attacks of porphyria are extremely rare before puberty and unusual after the menopause, having a peak occurrence in the third and fourth decades. Women are more frequently affected than men. In most patients, an attack will cease once the diagnosis has been made, appropriate treatment given and likely precipitants removed. However, particularly when there is delay in establishing the diagnosis, prolonged and very severe, life-threatening and sometimes fatal acute attacks may occur. Commonly ascribed precipitants include prescribed and illicit drugs, alcohol (particularly binge drinking), systemic infection and dieting either alone or in combination. In women, attacks may be related to the menstrual cycle, usually the luteal phase. However, in some cases no obvious aetiology is found. An acute attack of porphyria normally starts with continuous abdominal pain, which becomes progressively more severe and is associated with nausea, vomiting and constipation. The patient may also complain of pain in the lower back, buttocks and inner thighs. The abdominal pain is typically diffuse, with no localizing signs or evidence of an acute abdomen on examination. The severity of pain appears out of keeping with the physical signs and often requires the administration of large parenteral doses of opiates. In the absence of an obvious cause, this may trigger concerns about opiate dependence among clinical staff who have no previous experience of the condition. Diminishing pain, particularly where no treatment has been given, does not always herald the end of an acute attack, and patients should be carefully monitored for the development of neuropathy. The autonomic neuropathy that results in the gastrointestinal symptoms also gives rise to tachycardia and hypertension in approximately two-thirds of patients. Patients frequently become dehydrated during an acute attack and may also develop hyponatraemia, which may worsen if inappropriate volumes of hypotonic intravenous fluid are infused. Plasma sodium concentration can fall quickly to extremely low values, precipitating convulsions, which occur in about 5% of patients. Seizures may also occur as a neurological manifestation of the acute attack, and present a particularly difficult management problem as many antiepileptic drugs are implicated as a cause of acute attacks. Rhabdomyolysis, which may lead to renal failure, is a recognized but rare complication of an acute attack. Severe and untreated attacks can result in a predominantly motor peripheral neuropathy. In most cases, this is a symmetrical distal neuropathy resulting in wrist and foot drop, but occasionally there is progression to a flaccid paresis resembling Guillain–Barré syndrome and requiring prolonged ventilatory support. Prognosis for full recovery is excellent when attacks can be treated and halted. However, patients who require ventilatory support frequently experience a difficult recovery period, characterized by recurrent relapses triggered by the requirement for multiple prescribed drugs and complications such as infection. A small number of patients experience sensory changes such as dysaesthesia or hypoaesthesia in a similar distribution to the motor neuropathy. Other neurological signs reported include cranial nerve involvement including the optic nerve, which can lead to blindness. The CNS is frequently involved. Acute mental changes are common and include anxiety, insomnia, confusion, hallucinations and paranoia, which resolve completely following the acute attack. There is no evidence that any form of chronic psychiatric illness is associated with any of the acute porphyrias. Less common CNS features are cerebellar syndrome, pyramidal signs, transient cortical blindness and altered consciousness. Most patients have only one or a few attacks, with a major attack often being followed by two or three minor ones before full remission. However, about 5% of AIP patients suffer from frequent acute attacks, which in women may be premenstrual, and which may continue for several years (see p. 541). Obvious triggers are usually not found and the repeated admissions to hospital can severely affect the quality of life, particularly if patients have young families. Repeated admissions and high opiate requirement may also lead medical staff who are unfamiliar with the condition to question the diagnosis and the patient’s motives. Some patients develop a chronic pain syndrome. Pain is usually in the peripheries, often constant and may be triggered by minor stimuli. The cause of this is unknown; management is particularly difficult since it is important to avoid addictive analgesics. Treatment with haem arginate (p. 540) is usually of no benefit. Studies from several countries have demonstrated an increased risk of hepatocellular carcinoma (HCC) in the absence of chronic liver disease in patients with an acute hepatic porphyria even when this is clinically latent. The risk is particularly high in Sweden, where a recent study found a standardized increased risk ratio of 64 for AIP gene carriers aged over 54 years and recommended that AIP patients over the age of 50 years should be screened annually for HCC by ultrasound examination. The benefit from screening in other countries has yet to be assessed. It has also been suggested that all HCC patients in whom no obvious cause is found should be screened for acute porphyria. Renal impairment has been described as a complication of AIP, affecting particularly those patients who have previously suffered acute attacks. Hypertension is also common in these patients, but in many, the decline in renal function precedes the onset of hypertension, which may also be a consequence of the porphyria. Renal biopsies have shown glomerular sclerosis and interstitial fibrosis, but no evidence of inflammation or immunodeposits. A proportion of patients may progress to end-stage renal failure, requiring dialysis and/or renal transplantation. Patients who have had active disease should therefore have their renal function and blood pressure monitored regularly, and antihypertensive therapy should be instituted in an attempt to limit progression of renal impairment. Diagnosis of an acute attack in a newly presenting patient requires the demonstration of increased excretion of urinary PBG in a fresh, random sample of urine that has been protected from light. An assay capable of quantitative or semiquantitative measurement of PBG should ideally be available in all acute hospitals. In practice, many non-specialist units use rapid qualitative screening tests, particularly out of hours, which have low sensitivity and poor specificity. All positive screening tests should be confirmed by a specific quantitative method with expression of results in relation to creatinine to correct for urine concentration. If urine PBG and ALA concentrations are normal during the early phase of an acute illness, all acute porphyrias, including ADP, are excluded as a cause of that illness. However, both PBG and ALA may return to within normal limits within a few days after the onset of symptoms in VP and HCP, and negative findings should be interpreted with caution when there has been a delay in collecting samples. However, in both these disorders, urinary and faecal porphyrins remain elevated for a prolonged period. Conversely, urinary PBG excretion usually remains elevated for many months or even years after an acute attack in AIP, and an increased PBG does not necessarily indicate an acute attack unless a marked increase from baseline can be shown to coincide with symptoms. Management of a clinically diagnosed acute attack should be started immediately, without waiting for confirmation of a positive screening test or determination of the type of acute porphyria. Establishment of the type of acute porphyria requires analysis of plasma and faecal porphyrins (see Table 28.3; Fig. 28.2). Faecal porphyrin analysis is essential to distinguish HCP, in which coproporphyrin III accounts for most of the increased faecal porphyrin excretion, from AIP, in which faecal porphyrin excretion is normal or only slightly increased without any change in the coproporphyrin isomer ratio. Variegate porphyria can easily be identified by demonstrating a characteristic plasma porphyrin fluorescence emission peak at 625–628 nm. Enzyme analyses and genetic studies are not required for the diagnosis of new cases of clinically overt porphyria. Their use in family studies is detailed later in the chapter. Laboratory monitoring of patients during and after acute attacks by regular measurement of porphyrin precursors is rarely indicated; treatment should be guided by clinical assessment. The one exception is where a severe attack has progressed to flaccid paresis and clinical assessment is difficult. In these circumstances, weekly monitoring may provide useful information on whether the condition is stable or deteriorating. As soon as the acute attack is confirmed, drugs or other recognized precipitants should be withdrawn. Safe and effective management of symptoms and complications, with support from an expert centre, is essential to minimize the stress; effective pain relief is a major component. This invariably requires opiates, and support from a specialist pain team can be very helpful as very high doses may be required. A phenothiazine may be used for anxiety and restlessness, and to decrease the opiate requirements. Antiemetics should be prescribed for nausea and vomiting. Adequate fluid and energy intake should be ensured, if necessary by intravenous infusions of 0.9% sodium chloride containing a minimum of 5% dextrose, with regular monitoring of electrolyte status in view of the risk of hyponatraemia. Where drug treatment of precipitants such as infection, coexisting conditions or other features of an acute attack, such as hypertension, tachycardia or convulsions, are required, care should be taken to select medications that are considered safe. Information on drug safety is continually under review and safe drug lists are likely to change regularly (see Welsh Medicines Information Centre: www.wmic.wales.nhs.uk). Where no safe alternative is on the list, an expert centre should be consulted for further advice on patient management. Information about specialist centres offering support in Europe is available from the European Porphyria Network (EPNET): www.porphyria-europe.org.
The porphyrias
inherited disorders of haem synthesis
INTRODUCTION AND OVERVIEW

Biochemistry of haem synthesis
Overview of the porphyrias
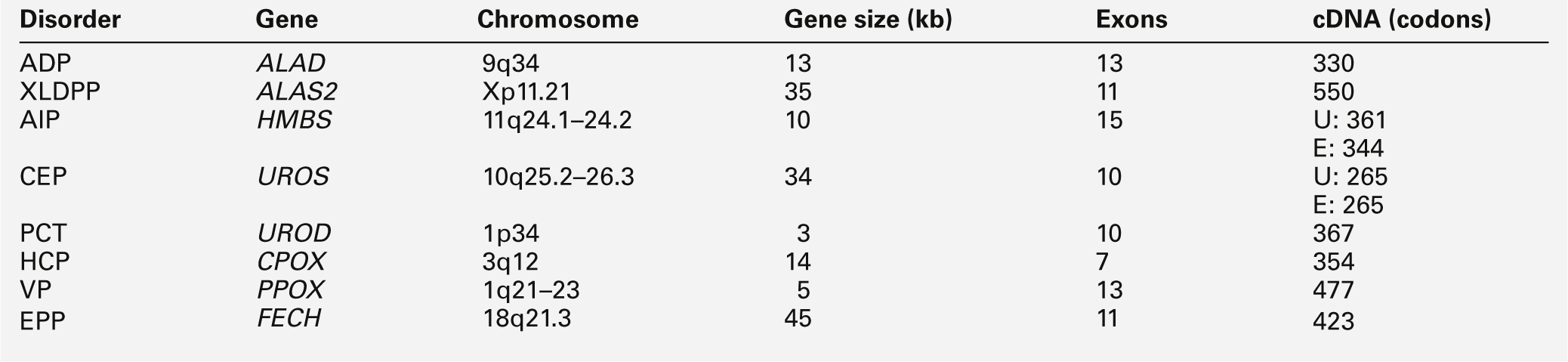
Molecular genetics of the porphyrias
PORPHYRIAS PRESENTING WITH ACUTE ATTACKS
The autosomal dominant acute porphyrias
Pathophysiology of acute attacks
Clinical presentation of acute attacks
Chronic complications
Diagnosis of acute porphyria
Management of an acute attack
Supportive treatment
Specific treatment

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


