CHAPTER 18 Miles J. Levy; Trevor A. Howlett CHAPTER OUTLINE CLINICAL ANATOMY OF THE PITUITARY AND HYPOTHALAMUS PHYSIOLOGY OF HYPOTHALAMO–PITUITARY–END ORGAN AXES CLINICAL ANATOMY AND PHYSIOLOGY OF THE ADRENALS ASSESSMENT OF NORMAL PITUITARY FUNCTION Dynamic tests of ACTH–adrenal function Cortisol normal ranges, borderline responses, assay precision and dynamic test reproducibility Assessment of growth hormone reserve Other tests of gonadotrophin secretion Dynamic tests of posterior pituitary function A clinical approach to assessment of the whole ACTH–adrenal axis Monitoring of pituitary function in disease states OTHER DIAGNOSTIC TECHNIQUES IN PITUITARY DISEASE PITUITARY HYPERSECRETION STATES Diagnosis and differential diagnosis of Cushing syndrome Thyroid stimulating hormone-secreting adenomas Gonadotrophin-secreting adenomas HYPOTHALAMIC AND PITUITARY DEFICIENCY STATES Diseases that may lead to generalized hypopituitarism Other isolated anterior pituitary deficiencies Clinical features of Addison disease Congenital adrenal hyperplasia Assessment of adrenal incidentaloma MONITORING PITUITARY AND ADRENAL REPLACEMENT THERAPY The investigation of disorders of the pituitary and hypothalamus and of adrenocortical function frequently causes great apprehension among non-endocrinologists, whether they are clinical biochemists or general physicians, because of their supposed complexity and the frequency of atypical, borderline or artefactual results. It is certainly true that the hypothalamo–pituitary axis plays the central role in the control of a large number of hormonal systems and of many other important aspects of homoeostasis. However, the clinically relevant physiology of each hormonal axis is well understood and the plasma concentrations of the majority of the hormones secreted by both the pituitary and the relevant peripheral endocrine glands can now be readily and accurately measured in most clinical biochemistry laboratories. Thus, a logical approach to the measurement of the relevant plasma hormone concentrations, applying basic physiological principles, can result in a relatively simple and efficient assessment of pituitary function in the majority of cases. This chapter will outline a clinically relevant and efficient approach to the assessment of pituitary and adrenal function. In many cases, simple measurement of basal, unstimulated plasma concentrations of the appropriate hormone(s) will give all the information required for clinical management. Some of the traditionally used dynamic endocrine tests can now be reasonably discarded, while others remain essential, and the choice of these will be discussed critically. Recommendations for clinical management of borderline or contradictory results will also be discussed. The pituitary gland (Fig. 18.1) lies within a bony compartment, the pituitary fossa or sella turcica, beneath the hypothalamus to which it is connected by the pituitary stalk. The fossa is separated from the subarachnoid space and cerebrospinal fluid by the diaphragma sellae. The adult human pituitary consists of two lobes (the third, neurointermediate lobe of other species is not present except in fetal life). The anterior lobe (adenohypophysis) is embryologically derived from cells of Rathke’s pouch. It contains a variety of cell types differentiated to secrete the majority of the peptide and glycopeptide hormones that control the function of peripheral endocrine organs: corticotrophs (adrenocorticotrophic hormone, ACTH), lactotrophs (prolactin, PRL), gonadotrophs (luteinizing hormone and follicle stimulating hormone: LH and FSH), thyrotrophs (thyroid stimulating hormone, TSH) and somatotrophs (somatotrophin or growth hormone, GH). The anterior lobe has no direct arterial blood supply but is supplied by the portal vessels that arise in the median eminence of the hypothalamus. The secretion of anterior pituitary hormones is controlled by releasing and inhibiting factors, which are predominantly peptides, released into the portal circulation from the nerve terminals of a variety of hypothalamic neurons. These neurons in turn are subject to the effects of the modulatory neurotransmitters of other neurons, within the hypothalamus and beyond. FIGURE 18.1 Functional anatomy of the human pituitary and hypothalamus. The critical role of the portal circulation in the control of anterior pituitary function means that any disease process that interferes with this blood supply, for example non-functioning tumours of the pituitary or hypothalamus, will often result in severe pituitary dysfunction, even if the actual hormone-secreting cells of the anterior pituitary have not themselves been destroyed. The posterior lobe (neurohypophysis), in contrast, is embryologically derived from the brain, has a direct arterial blood supply and is not controlled via a portal circulation. Hormonal secretion occurs directly from the nerve terminals of vasopressin (antidiuretic hormone, ADH) and oxytocin neurons, whose cell bodies lie within the hypothalamus, primarily in the supraoptic and paraventricular nuclei. Both the anterior and posterior lobes contain a variety of other cell types. These are not directly responsible for hormonal secretion into the peripheral circulation, although they may have important modulatory or paracrine roles in the control of pituitary function. Many other important neural structures lie within or adjacent to the hypothalamus, notably the optic chiasm and the neural centres controlling thirst, osmoregulation, appetite, temperature homoeostasis and circadian rhythm. Diseases of the pituitary and hypothalamus can result in dysfunction of any of these structures, in addition to the hormonal syndromes described here. In particular, visual field loss due to compression of the optic chiasm is a frequent finding with large pituitary tumours. These anatomical relationships indicate that the pituitary gland is truly at the interface of the mind and body, with a central role in homoeostasis, explaining its source of fascination for physiologist and physician alike. The basic physiology of the important endocrine axes is summarized diagrammatically in Figures 18.2 and 18.3. Many additional releasing, inhibiting and modulating factors have been identified, primarily in animal experiments, but none of these has yet been shown to be of relevance clinically, nor for the understanding of the principles of pituitary function testing. Hormones that can be measured in the laboratory are indicated in the figures. Peripheral plasma concentrations of hypothalamic releasing factors can also be measured (with difficulty), but for the most part do not reflect hypothalamic activity and are not of clinical relevance. FIGURE 18.2 Physiological relationships of the anterior pituitary. Hormones (in bold) can be readily measured in most clinical biochemistry laboratories. See text for explanation of abbreviations. FIGURE 18.3 Physiological relationships of the posterior pituitary. Osmolalities (italicized) can be readily measured in most clinical biochemistry laboratories. Hormones (emboldened) can be measured with difficulty in a few centres. Adrenocorticotrophic hormone (ACTH) stimulates adrenal cortisol secretion and is itself controlled by hypothalamic corticotrophin releasing factor (CRF). Corticotrophin releasing factor is now known to be a complex of factors including a 41-residue peptide (CRF-41 or corticotrophin releasing hormone, CRH) and vasopressin, which act synergistically. Adrenocorticotrophic hormone, and therefore cortisol, are secreted with a pronounced circadian rhythm: concentrations are low, typically undetectable, at midnight (if asleep), rise in the final hours of sleep to reach a peak shortly after wakening in the morning and steadily decline throughout the rest of the day with superimposed peaks of secretion with meals, exercise and stressful events. ‘Stress’ (‘fight and flight’, illness, hypoglycaemia, surgery etc.) is a major determinant of ACTH and cortisol secretion, which therefore means that basal concentrations in a well individual may not always reflect the ability to respond to illness, and conversely that high concentrations in an individual with other significant disease do not always imply oversecretion. Secretion of growth hormone (GH) is controlled by complementary stimulatory and inhibitory factors, GH releasing hormone (GHRH, a 44-residue peptide) and somatostatin (SMS, a cyclic, 14-residue peptide), respectively. Growth hormone secretion occurs in pulses, predominantly at night during the early hours of sleep. Only infrequent, small pulses occur during the day when plasma concentrations are usually undetectable, although GH secretion also occurs in response to stress. Growth hormone’s effects on growth are mediated via synthesis of insulin-like growth factor 1 (IGF-1 – previously called somatomedin C), which is synthesized mostly in the liver but also in peripheral tissues. Prolactin secretion is unique in being predominantly controlled by an inhibitory hypothalamic factor, dopamine. This fact explains why PRL deficiency is rare and why any disease that interferes with pituitary anatomy, and thus the flow of portal blood, may be associated with hyperprolactinaemia. Plasma PRL is raised physiologically during pregnancy and lactation, and release also occurs in response to stress. The physiological role of PRL in non-lactating women and in men is unknown. Hypothalamic thyrotrophin releasing hormone (TRH), a tripeptide, controls TSH secretion, which in turn stimulates the synthesis and secretion of thyroid hormones thyroxine (T4) and tri-iodothyronine (T3). Secretion is regulated primarily to maintain normal circulating concentrations of thyroid hormones, although there is also a slight circadian rhythm. Luteinizing hormone and FSH are both controlled by a single stimulatory decapeptide, gonadotrophin releasing hormone (GnRH or luteinizing hormone releasing hormone, LHRH). The major role of LH in the male is to stimulate testosterone secretion by testicular Leydig cells, and in the female to stimulate ovulation at mid-cycle. Follicle stimulating hormone controls spermatogenesis in the male and ovarian follicular development, and therefore oestradiol secretion, in the female, and in both sexes stimulates gonadal secretion of inhibin, which is responsible for some negative feedback control. The most important feature of the secretion of GnRH, and thus LH and FSH, is its pulsatile nature, imposed by the hypothalamic pulse generator. It is likely that differential secretion of LH and FSH is achieved by modulation of the frequency and amplitude of pulsatile GnRH secretion, and the peripheral effects of LH and FSH are critically dependent on appropriate pulsatile variation in concentrations. Gonadotrophin releasing hormone pulses occur, on average, every 90 min, but in the female are more rapid in the follicular phase and slower in the luteal phase of the menstrual cycle. During early puberty, the majority of pulses occur at night, during sleep. Vasopressin (antidiuretic hormone, ADH) secretion is controlled primarily by osmotic and blood volume sensing mechanisms (osmoreceptors and baroreceptors, respectively). High plasma osmolality and hypovolaemia or hypotension are the major stimulatory factors. Vasopressin acts directly on the kidney to increase water reabsorption in the renal collecting ducts and thus reduce urine volume. Corticosteroids are necessary for the excretion of free water, so deficiency may mask the symptoms of ADH deficiency. Oxytocin is released during labour and during suckling, but mechanisms controlling its secretion have not been studied extensively in humans and there appears to be no clinical syndrome related to oxytocin deficiency. Secretion of all pituitary hormones is controlled, to a greater or lesser extent, by negative feedback. This occurs at all levels, including the direct inhibition by a hormone of its own secretion (‘ultra-short-loop’), but the most important negative effects are generally those of the peripheral ‘end hormone’ on hypothalamic and/or pituitary secretion. The existence of such negative feedback is of crucial importance to the interpretation of basal hormone concentrations in pituitary disease and is the basis of a number of dynamic tests of pituitary function. The two adrenal glands lie superior to the kidneys and are composed of the inner catecholamine-secreting medulla, which is controlled centrally by direct innervation, and the outer cortex, which is controlled by classic endocrine pathways. Glucocorticoid and androgen secretion is predominantly from the zona fasciculata and zona reticularis and controlled by pituitary ACTH secretion, while mineralocorticoid secretion is predominantly from the zona glomerulosa and controlled via the renin–angiotensin system (see Chapter 4). This means that the patterns of adrenal deficiency vary with aetiology: thus a disease that destroys the adrenal itself (e.g. autoimmune Addison disease, adrenal tuberculosis or bilateral adrenalectomy) results in deficiency of both glucocorticoid and mineralocorticoid secretion, whereas ACTH deficiency from pituitary disease results in glucocorticoid deficiency alone and, thus, less marked disturbances of salt and water balance. Even complete adrenalectomy does not seem to cause a clinical deficiency of catecholamines, since secretion by the rest of the sympathetic nervous system appears to compensate for the loss of adrenal secretion. Adrenal steroid synthesis involves a complex and intersecting sequence of enzymatic steps within the cytoplasm and mitochondria of the adrenal cell. Pathways are illustrated in Figure 18.4. The initial, rate-limiting, step in steroidogenesis in the normal subject is the conversion of cholesterol to pregnenolone. Adrenocorticotrophic hormone has rapid effects on cholesterol transport into mitochondria and longer-term effects on transcription of genes encoding a number of enzymes in the synthetic pathway. These pathways are of clinical significance in congenital adrenal hyperplasia, where inherited defects in a variety of these enzymes give rise to the various subtypes and clinical syndromes (see Table 18.1), and in clinical pharmacology, since metyrapone, a drug that inhibits 11β-hydroxylase, can be used to decrease cortisol synthesis in Cushing syndrome. FIGURE 18.4 (A) The pathways of steroid hormone synthesis within the adrenals (and gonads). Enzymes are named by reaction catalysed (e.g. 21-hydroxylase) or by enzyme name (e.g. p450C11). p450 enzymes are in mitochondria and other enzymes are in the cytoplasm; steroid synthesis involves repeated passage of precursors between these cellular compartments. (B) Basic structure of the steroid molecule (from Kumar and Clark 2012 Clinical medicine. 8th ed. Edinburgh: Churchill Livingstone, with permission). TABLE 18.1 Subtypes of congenital adrenal hyperplasia A single, basal blood sample, especially if taken at 09.00 h, can give extensive and, for some purposes, complete information regarding pituitary function. The blood should ideally be taken under resting, unstressed conditions but, while the stress of major illness or surgery significantly alters results and thus might cause diagnostic errors when investigating pituitary hyperfunction, minor anxiety regarding hospital attendance or venepuncture probably has little effect. The pituitary–thyroid axis is fully assessed by the simultaneous measurement of plasma free thyroid hormone concentrations and TSH. Thyroid stimulating hormone deficiency is characterized by a low plasma free T4 concentration without the expected elevation of plasma TSH. In hypopituitarism, TSH may be low (although rarely undetectable) or within the normal range (inappropriately for the low T4), which represents the most potent argument against the use of TSH as the sole screening test for thyroid function in a laboratory. Basal plasma PRL gives full physiological information regarding the function of its axis. Concentrations frequently vary from day to day (particularly minor elevations), so that 2–3 basal samples are usually necessary before any treatment is instituted. In the male, a normal basal plasma testosterone (preferably taken at 09.00 h, in view of the significant circadian rhythm) effectively demonstrates normal LH secretion, and demonstration of a normal sperm count would confirm normal FSH secretion (although this is rarely performed unless infertility is an issue). Gonadotrophin deficiency is diagnosed by the combination of a low plasma testosterone with low or inappropriately ‘normal’ (rather than elevated) concentrations of LH and FSH. The significance of borderline testosterone concentrations – near or just below the lower end of the reference range – together with normal LH and FSH, particularly in the ageing male, is an area of growing clinical interest but still with considerable controversy and is discussed more fully later in this chapter. Assessment of the pituitary–gonadal axis in the female is complicated by the physiological changes during the menstrual cycle. A normal luteal phase plasma progesterone is the ultimate biochemical test of the adequacy of the entire axis. Spontaneous, normal, regular menstruation also excludes gonadotrophin deficiency without the need for additional biochemical tests. Gonadotrophin deficiency is suggested in the context of amenorrhoea by the presence of a consistently low plasma oestradiol in the absence of elevation of plasma LH and FSH, although a variety of ‘hypothalamic’ causes of amenorrhoea also show this combination (see below). In postmenopausal women (or, in the context of longstanding amenorrhoea, arbitrarily women aged >50 years), who are not on oestrogen replacement, the simple absence of the usual menopausal elevation of LH and FSH is sufficient to diagnose gonadotrophin deficiency. Basal plasma cortisol is not always sufficient to diagnose ACTH deficiency or normality, because of the circadian rhythm and since concentrations that need to be achieved during physiological stress are much higher than those found under normal basal circumstances. Nevertheless, a basal cortisol >550 nmol/L in an unstressed individual certainly excludes ACTH deficiency and, in our experience, a 09.00 h cortisol <100 nmol/L is never, and >400 nmol/L is always, associated with a normal ACTH reserve (see below). Basal concentrations of GH are usually unhelpful in the diagnosis of deficiency, since basal concentrations in normal individuals are usually undetectable. A low serum IGF-1 (using age-related normal ranges) is a good predictor of severe GH deficiency but, conversely, an IGF-1 in the normal range does not exclude severe deficiency that might respond to replacement therapy. Plasma vasopressin and oxytocin concentrations are measured by only a few centres and are not used routinely in the assessment of basal pituitary function. Measurement of paired plasma and urine osmolality on a basal early morning sample does, however, give valuable information. A urine osmolality >600 mmol/kg excludes diabetes insipidus (DI) as long as plasma osmolality is normal (280–295 mmol/kg). Conversely, DI is suggested by an elevated plasma osmolality (>300 mmol/kg) with a urine osmolality <600 mmol/kg. Other combinations are non-diagnostic and require a water deprivation test if DI is suspected clinically. Oxytocin secretion is never investigated clinically. The order of progression of hypopituitarism with progressively severe pituitary disease is usually predictable. Growth hormone and gonadotrophin secretion are usually most susceptible to damage, followed by ACTH and TSH, with PRL deficiency occurring only rarely and usually only after extensive pituitary damage by disease or surgery. It is thus unusual, although not impossible, for a patient with regular menstruation or normal male gonadal function to develop ACTH deficiency first in the course of their disease. Diabetes insipidus usually only occurs with inflammatory or invasive pituitary disease, after surgery or with diseases primarily involving the hypothalamus. Thus clinical background may influence the degree of investigation considered appropriate in an individual patient with borderline or equivocal results in a single axis. The insulin stress test (IST) (or insulin tolerance test, ITT) has traditionally been the ‘gold standard’ test to assess the adequacy of the ACTH–adrenal axis. After adequate hypoglycaemia (blood glucose <2.2 mmol/L), a normal rise of plasma cortisol (traditionally to >550 nmol/L) excludes ACTH and cortisol deficiency. Patients with such a response are capable of producing a normal adrenocortical response to stressful illness, without the need for replacement therapy. The increment of plasma cortisol does not add additional information. Patients with borderline peak responses (400–550 nmol/L) may only require steroid replacement during stressful illnesses, but patients with lower responses will typically be symptomatic and almost always require standard replacement therapy. An IST is contraindicated by cardiac disease (an ECG should be normal) and by epilepsy (or other unexplained blackouts) and always requires close medical and nursing supervision to maintain safety. For this reason, alternative, simpler tests have been sought, although none has yet achieved universal acceptance. A retrospective audit of 230 ISTs in our own laboratory indicated that patients with a basal cortisol <100 nmol/L never, and >400 nmol/L always, demonstrated a normal cortisol response, so that an IST is probably unnecessary in such patients (representing 50% of ISTs performed in our study) if the only aim is to assess pituitary–adrenal reserve. In our experience, patients who have recently discontinued steroid replacement with a basal cortisol <200 nmol/L also never have a normal response. This test, involving administration of tetracosactide (synthetic ACTH1–24), is the cornerstone for diagnosis of primary adrenal failure, but is increasingly used in the context of pituitary disease. It has long been accepted that a normal cortisol response to tetracosactide excludes Addison disease, but to test for ACTH deficiency by administration of ACTH is clearly illogical on purely physiological grounds, since it directly tests only the adrenal response and not that of the pituitary or higher centres. However, presumably since ACTH deficiency leads to adrenal atrophy, a number of workers have shown close correlations between the cortisol responses to a simple intravenous tetracosactide test and to an IST, and have suggested that the former is a satisfactory test of the ACTH–adrenal axis as long as the test is not done within four weeks of the pituitary insult. A majority of UK endocrinologists now accept that a normal plasma cortisol response, 30 min after tetracosactide (250 μg), adequately excludes ACTH deficiency, and several long-term clinical observational studies have confirmed the safety of such an approach. This test was originally validated with i.v. tetracosactide but responses to i.m. injection are equivalent. However, normal responses are different (typically 150 nmol/L or so higher) at 60 min, so an appropriate normal range is needed for each time point and, in practice, we recommend only measuring the 30 min value to avoid confusion. Since the standard 250 μg dose of tetracosactide is substantially supraphysiological, many workers have advocated use of a lower dose stimulation test (typically 1 μg). However, this approach is more complicated since no suitable dose preparation exists, and it has not been demonstrated to be significantly superior in clinical practice to the traditional test. The glucagon test (1 mg i.m.) has been advocated to assess ACTH and GH reserve when an IST is contraindicated (usually with the same criteria for a normal response). Glucagon frequently causes nausea and vomiting, normal individuals sometimes fail to show a ‘normal’ response and sampling must continue over a 4 h period, making this test relatively unattractive as a routine alternative. The metyrapone test (750 mg orally, four-hourly for 24 h) has been widely used as a test of ACTH reserve, particularly in North America; it measures the response of urinary cortisol precursors or, more recently, plasma 11-deoxycortisol, to blockade of adrenal cortisol synthesis by this drug. The test is time consuming, often unpleasant for the patient and does not test the response to physiological stress. An intravenous short tetracosactide test is now recognized by most endocrinologists to be a more appropriate and simpler alternative to either of these tests, unless information on GH reserve is also required. Normal ranges for cortisol, particularly for dynamic tests, were predominantly developed and validated using old assay technologies (e.g. fluorimetry), which are no longer in use, and although a variety of adjustments (typically lowering of cortisol cut-off concentrations for a normal response) have been suggested, these have rarely been completely validated. Detailed studies of normal responses to tetracosactide have shown substantial differences with gender and between commonly used cortisol assays (see Clark et al. in Further reading, below). Most workers and most assays report a lower limit of the normal response between 500 and 600 nmol/L, but some other published studies report lower peak responses than this in some apparently healthy individuals (lowest values ~400 nmol/L). Similarly, some recent studies reporting cortisol responses to IST in healthy volunteers have reported peak responses in some individuals on some occasions between 400 and 500 nmol/L. Studies of test–re-test variability for both the tetracosactide and IST tests have mostly reported a coefficient of variation (CV) of around 10%. External quality control schemes also frequently report an interassay/intermethod CV for cortisol of 10% or higher. Therefore, if we assume conservatively that the combined variability from these two sources is represented by an overall CV of 10%, then simple statistics show that the 95% confidence limits for a test response with a ‘true’ value of 500 nmol/L actually extends from 400 to 600 nmol/L. These considerations potentially apply to both the tetracosactide test and the IST. All of these factors lead the authors to define basal cortisol and cortisol responses to tetracosactide and other dynamic tests broadly rather than using a single precise cut-off, and to recognize a group with borderline or equivocal responses who cannot be clearly classified as ‘normal’ or ‘abnormal’: • 400 nmol/L: a basal cortisol above this value indicates a normal hypothalamo–pituitary–adrenal (HPA) axis – unless the patient is acutely unwell or has ongoing unexplained symptoms consistent with hypoadrenalism. A cortisol response to tetracosactide or IST below this concentration usually means that routine steroid replacement is required • 600 nmol/L: a cortisol response to tetracosactide to above this concentration at 30 min almost always means that ACTH deficiency or primary adrenal failure is excluded. A cortisol response to above this concentration in an IST certainly excludes the need for steroid replacement even during severe intercurrent illness • 100–400 nmol/L: basal cortisol concentrations in this range need a dynamic test to confirm the abnormality of the axis; a tetracosactide or IST peak response in this range usually indicates a requirement for routine steroid replacement • 400–600 nmol/L: peak responses to tetracosactide or IST in this range in patients with known pituitary disease may not require the patient to receive regular replacement, but do require education of the patient about the possible need for replacement and symptoms of hypoadrenalism during intercurrent illness (and often provision of an emergency supply of hydrocortisone). Responses to tetracosactide in this range can sometimes be seen in normal individuals, so if there is no other evidence of adrenal or pituitary disease, further investigation may be required (e.g. renin and ACTH concentrations, possibly followed by IST to exclude isolated ACTH deficiency if the former are normal). Interpretation of isolated responses in this range are further complicated by the fact that patients with chronic fatigue syndrome may show borderline reduced cortisol dynamic responses, although in most cases they do not respond symptomatically to steroid replacement. There are several biochemical tests that are used in clinical practice to confirm growth hormone deficiency (GHD). The clinical features of GHD are non-specific, and the different GH stimulation tests have different degrees of ability to stimulate GH release. For this reason, there continues to be debate about the most appropriate test and what extent of GH response constitutes true deficiency. The insulin stress test (IST) remains the gold standard test of GH deficiency. In adults, a peak GH response to adequate hypoglycaemia (serum glucose <2.2 mmol/L) of >7 μg/L indicates a normal GH reserve, whilst a peak GH <3 μg/L, as defined by the UK National Institute for Health and Care Excellence (NICE), indicates severe deficiency. In children, a peak GH <6.7 μg/L after two provocative tests supports a diagnosis of growth hormone deficiency, although only one dynamic test is required in children with known hypothalamo–pituitary disease. The IST is unpleasant for patients and requires close medical supervision as it is potentially hazardous and is contraindicated in ischaemic heart disease and epilepsy. As well as being a reliable GH stimulus, the IST is also a good assessment of ACTH reserve so it a useful way of assessing GH and ACTH reserve simultaneously. Children of both sexes may show subnormal GH responses to hypoglycaemia (and other dynamic tests) in the years immediately preceding puberty (objectively when the bone age is > 10 years). At the present time, there is no consensus with respect to the practice of ‘priming’ with gonadal steroids prior to such tests. The glucagon test can be useful in the assessment of GHD when an IST is contraindicated, but it is a less reliable stimulus of GH and a poorer test of ACTH reserve than the IST, and its mechanism of action is not well understood. The criteria for diagnosing GHD in the glucagon test are the same as for the IST described above. Other provocative tests of GH reserve include the GH-releasing hormone (GHRH) + arginine and GHRH + GH-releasing hexapeptide (GHRP-6) test, both of which have now been evaluated in large numbers of patients. The GHRH + arginine test may be particularly useful for young patients with radiation-induced GHD as occasionally the IST can give false negative results. Because GHRH + arginine is a more powerful GH stimulus, the cut-off point for diagnosing GHD is <9 μg/L. In an attempt to avoid an IST, a variety of other pharmacological tests of GH reserve have also been used, particularly in children, including clonidine (0.15 mg/m2) and arginine infusion (0.5 g/kg). Use of these tests tends to be a matter of local preference; all are associated with rather variable responses and the peak GH values used to diagnose deficiency vary from laboratory to laboratory. Growth hormone is released during exercise and some laboratories have used an exercise test to assess GH reserve. Intensive, well controlled exercise (bicycle ergometer or treadmill) may exclude GH deficiency (by a peak GH response >6.7 μg/L), but less well controlled exercise (‘run up and down the stairs’) is unreliable. Because GH is secreted in a pulsatile fashion with increased pulses at night, serial blood tests, which can be done overnight or over 24 h, can be used to monitor the frequency and amplitude of GH pulses. Because this approach is labour intensive, requiring hospital admission with multiple blood samples, it is mainly a research tool. Measurement of urinary GH is not part of routine clinical practice because of its low concentration in urine, but sensitive assays may make this possible in the future, particularly for the surveillance of GH replacement therapy. Insulin-like growth factor 1 is produced by the liver in response to GH and, although measurement of its concentration is a useful diagnostic adjunct to provocative GH stimulation tests, it should not be used alone to diagnose GHD. However, the finding of a low IGF-1 in patients with three or four other pituitary deficiencies makes the likelihood of GHD sufficiently high that a formal provocative test may not be necessary. In patients with isolated GHD that developed in childhood, it can be difficult to know whether GH replacement should be continued into adulthood. Patients with genetic hypopituitarism or multiple hormone deficiencies are highly likely to need to continue GH replacement. However, it may be able to be discontinued in patients with isolated or less severe GHD. In this situation, patients are re-tested during the period of transition between childhood and adulthood and the current consensus guidelines suggest a cut-off of <6 μg/L during an IST to indicate a continuing need for GH replacement. In this situation, the presence of clinical symptoms can also help the decision about whether to continue GH, which is best done by evaluation of the Adult Growth Hormone Deficiency Assessment (AGHDA) score. Consensus guidelines suggest that young adults with GHD should continue GH until the age of 25 years, when peak bone mass is likely to have been achieved. Thyrotrophin releasing hormone (200 μg i.v.) and LHRH (100 μg i.v.) tests have been used traditionally in combination with the IST to assess pituitary reserve in the combined pituitary function test. Growth hormone releasing hormone testing (100 μg i.v.) and CRH testing (100 μg i.v.) have also more recently been advocated as additional tests. All these releasing hormone tests assess only the readily releasable pituitary pool of their relevant hormones and do not assess the ability of the axis to respond to appropriate physiological stimuli. When the cause of hypopituitarism is at the hypothalamic level (which is frequently the case), then ‘normal’ TRH, LHRH, GHRH or CRH responses may be seen in the presence of deficiency documented by other means. Conversely, ‘subnormal’ responses are often seen when the axis is otherwise felt to be normal. Furthermore, a ‘delayed’ TRH test response (peak TSH at 60 min rather than 20 min), initially considered to be characteristic of hypothalamic disease, is not infrequently seen in primary pituitary disease. For these reasons, the authors believe that all these releasing hormone tests should now play no part in routine pituitary function testing, but be reserved (if used at all) for the rare cases requiring differentiation between pituitary and hypothalamic deficiency states. When the differentiation of gonadotrophin deficiency from other causes of ‘hypothalamic amenorrhoea’ is in doubt, a clomifene test (3 mg/kg, maximum 200 mg, daily for seven days) may give additional information. Patients with organic gonadotrophin deficiency show no increase in LH and FSH (measured on days 0, 4, 7 and 10), whereas some patients with hypothalamic amenorrhoea, weight-related amenorrhoea or delayed puberty show a gonadotrophin rise, usually followed by menstruation (about 28 days later). In early puberty, LH and FSH may show a characteristic fall rather than a rise. Measurement of plasma LH every 10–15 min for 6–8 h or more, when analysed by an appropriate computer algorithm, allows detailed assessment of LH pulse frequency and amplitude. This may give considerable insight into pituitary physiology but, in view of the time and number of assays involved, is essentially a research tool. Water deprivation is the standard physiological test of vasopressin secretion. The patient is observed, without access to water, with serial measurements of plasma and urine osmolality, urine volumes and body weight over a period of up to 8 h. If only borderline deficiency is suspected then it is wise, in order to shorten the procedure, to ask the patient to take no fluids from midnight before a morning test; otherwise the patient should be allowed free fluids prior to the test to avoid severe dehydration. Simple measurement of 24 h urine volume prior to the test may guide which approach is most appropriate; if this is <2 L/24 h then DI is unlikely in the first place. If body weight falls by more than 3% during the test, this suggests severe dehydration. The test should be stopped, and plasma osmolality measured for confirmation before allowing access to fluids. Normal vasopressin secretion is demonstrated by a urine:plasma osmolality ratio of >2:1 (or, more simply, a urine osmolality >600 mmol/kg), in the presence of a normal plasma osmolality (280–295 mmol/kg). A high plasma osmolality (>300 mmol/kg) with a urine osmolality of <600 mmol/kg is diagnostic of DI. Intermediate responses are non-diagnostic and water deprivation must be continued or repeated, until diagnostic values are obtained. Once a diagnostic response has been obtained, then desmopressin (2 μg i.m.) is administered, free fluids are allowed and urinary osmolality is measured hourly for 2 h. In the presence of DI, full concentration of the urine after desmopressin indicates vasopressin deficiency rather than nephrogenic DI. Patients with primary polydipsia frequently commence the test with low urine and plasma osmolalities due to water overload, and may fail to concentrate the urine adequately after the standard period of water deprivation since the plasma osmolality is frequently only increased into the low–normal range during this time. The renal response to desmopressin may also be impaired under these circumstances due to ‘washout’ of the renal concentrating gradients. Severe DI can be diagnosed easily by a water deprivation test, but mild or borderline DI, and other more subtle defects of osmoregulation and thirst, may lead to equivocal findings despite protracted water deprivation. Under such circumstances, a hypertonic saline infusion (5% NaCl infused at 0.06 mL/kg per min for 120 min) may be used to delineate the pathology more accurately. Plasma osmolality and vasopressin are measured every 30 min but, as vasopressin measurements are not performed in most laboratories, this test is often only carried out in specialist centres. Discussion of the interpretation of this test is beyond the scope of this chapter (see Chapter 4 and Baylis, in Further reading, below). The multiple tests available and the controversies regarding their use and interpretation mean that the detail in which pituitary function is investigated remains a matter for clinical judgement. The authors propose the following protocol for the initial assessment of a patient with suspected pituitary disease. 1. Measure basal pituitary function on a single 09.00 h sample (see Appendix 18.1, p. 371). 2.
Hypothalamic, pituitary and adrenal disorders
INTRODUCTION
CLINICAL ANATOMY OF THE PITUITARY AND HYPOTHALAMUS
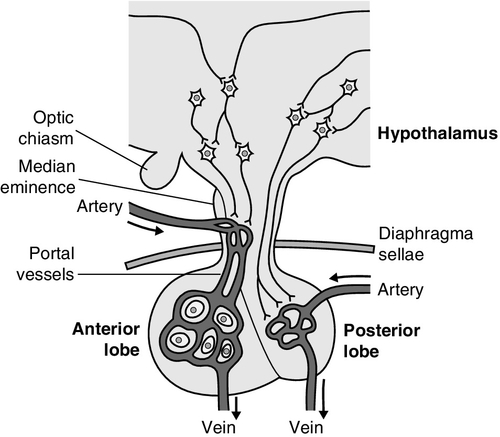
PHYSIOLOGY OF HYPOTHALAMO– PITUITARY–END ORGAN AXES
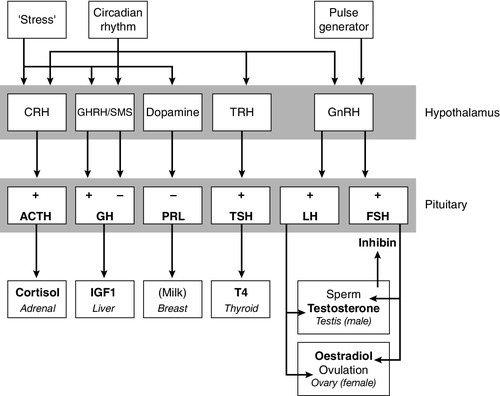
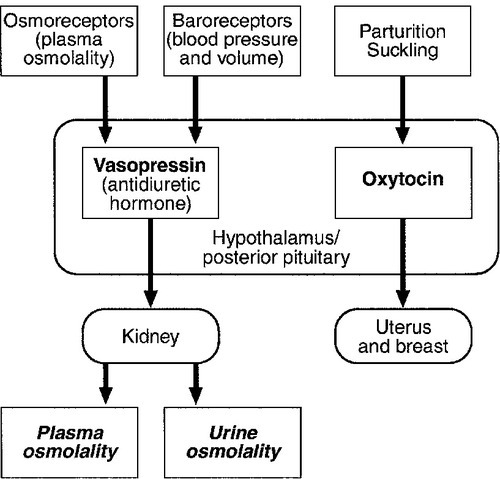
CLINICAL ANATOMY AND PHYSIOLOGY OF THE ADRENALS
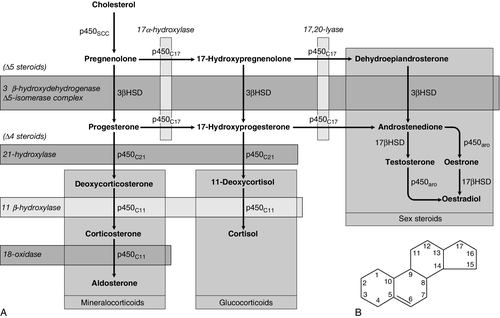
Enzyme defect
Frequency
Clinical features
21-hydroxylase p450C21/CYP21
95% of cases
1 in 15 000 live births
Ambiguous genitalia in females
Virilization, some salt losing
11-β-hydroxylase p450C11/CYP11B
< 5% of cases
Ambiguous genitalia in females
Virilization, hypertension
17-α-hydroxylase p450C17
Rare
Ambiguous genitalia and lack of virilization in males
Hypertension
3β-hydroxysteroid dehydrogenase 3βHSD/HSD3B2
Rare
Ambiguous genitalia in males
Mild virilization in females
Occasional Addisonian crises
Cholesterol desmolase p450SCC/CYP11A
Rare
Addisonian crises
ASSESSMENT OF NORMAL PITUITARY FUNCTION
Basal hormonal investigations
Dynamic tests of ACTH–adrenal function
Insulin stress test
Short tetracosactide (synacthen, tetracosactrin, ACTH) test
Other tests
Cortisol normal ranges, borderline responses, assay precision and dynamic test reproducibility
Assessment of growth hormone reserve
Insulin stress test
Other pharmacological tests
Exercise testing
Assessment of physiological growth hormone secretion
Re-evaluation of GH status in young adults
Releasing hormone tests
Other tests of gonadotrophin secretion
Clomifene test
Assessment of luteinizing hormone pulsatility
Dynamic tests of posterior pituitary function
Water deprivation test
Hypertonic saline infusion
Summary
Outline protocol for the investigation of a patient with pituitary disease
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


