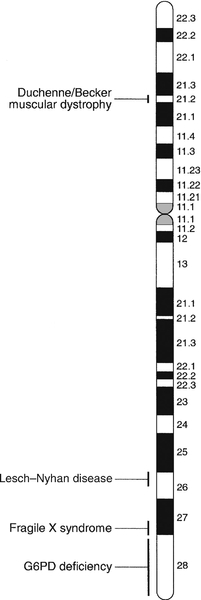
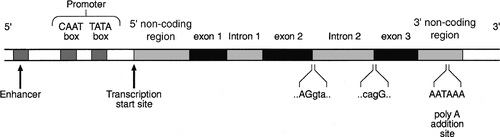
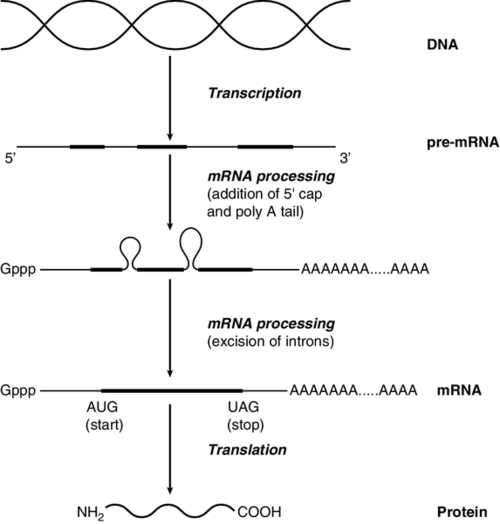
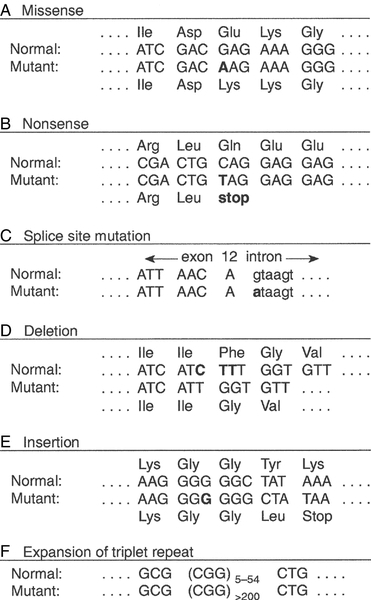
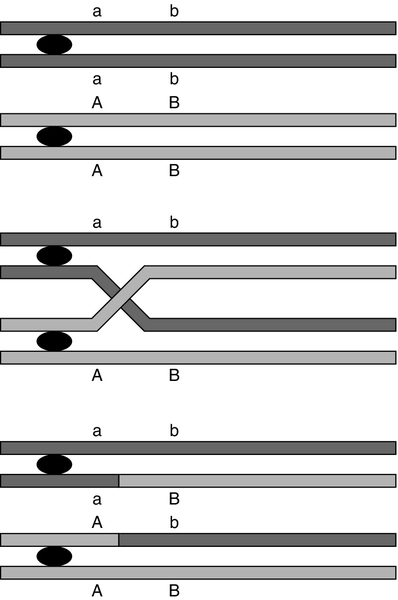
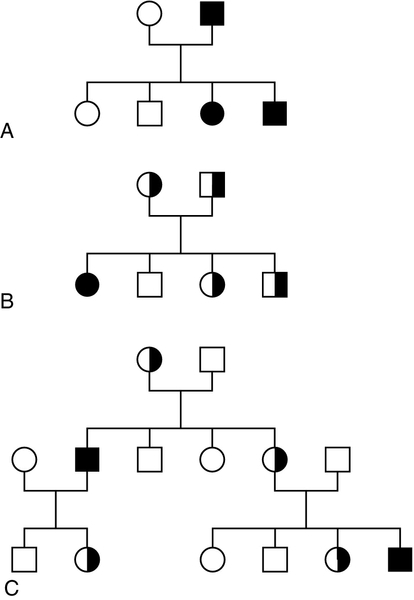
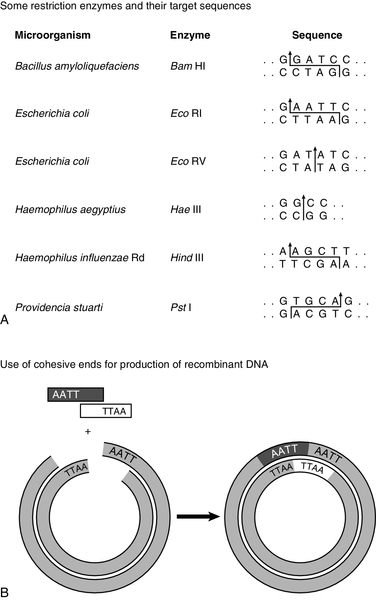
CHAPTER 43 CHAPTER OUTLINE Mutation, the source of diversity and disease Genesis of an individual: the formation of gametes Genes in families and populations The variable expression of genetic disease THE TECHNIQUES OF GENETIC ANALYSIS Detection of specific sequences in DNA THE APPLICATIONS OF DNA ANALYSIS Inherited diseases – some examples Multifactorial and polygenic disease If we distinguish the actual combination of genes possessed by an individual, that is the genotype, from the observable activity of those genes, the phenotype, then study of inherited disease in clinical biochemistry laboratories has traditionally been concerned exclusively with analysis of phenotypes. The last two decades have witnessed a dramatic change in this situation: molecular biological techniques are now a much more common part of the repertoire of clinical biochemistry laboratories. Initially, the identification of genes responsible for inherited diseases involved heroic efforts, requiring expensive, complex and extremely time-consuming procedures plus a few strokes of luck. Once a gene has been identified, however, modern analytical techniques make detection of mutations more straightforward than before. Although each nucleated human cell contains about two metres of deoxyribonucleic acid (DNA) (around 3 billion bases), it is the fundamental simplicity of DNA – its building blocks comprise just four nucleotides – that favours its automated analysis. With a few exceptions, every cell in the body of an individual contains a complete copy of their DNA (or genome). For this reason, genetic analysis can be carried out on almost any nucleated cell type (such as lymphocytes or buccal mucosal cells) that can conveniently be collected. The application of DNA analysis now extends well beyond diagnosis of the classic inherited diseases to include, for example, the diagnosis and prognosis of cancer. This chapter provides a general background to clinical laboratory applications of molecular genetic analysis. The emphasis is on diagnostic techniques with the potential for automation, utilizing polymerase chain reactions (PCRs), since classic techniques such as Southern blot analysis are not widely used in hospital biochemistry laboratories, but tend to be restricted to specialist molecular genetics departments. As far as possible, only a very basic knowledge of molecular biology has been assumed, but several excellent introductions to the topic are available (see Further reading) and a glossary is provided on page 872. A common working definition is that a gene is a sequence of nucleotide bases in DNA that codes for a single polypeptide, but the complexity of genomic organization is such that it is probably unwise to adhere rigidly to any one definition of the gene. Towards the end of the 19th century, it was already accepted that linear groups of ‘invisible self-propagating vital units’ were present in chromosomes. Mendel’s discovery (1865) that inheritance is particulate was rediscovered and publicized at the beginning of the 20th century, and the term gene was introduced to describe Mendel’s ‘particulate elements’ in 1909. By 1911, a specific gene (for colour blindness) had already been assigned to a particular chromosome (the X chromosome). With the work of Garrod, who first presented his studies on alkaptonuria in 1902, the association of specific diseases with inherited Mendelian traits became established. Certain stains produce clearly defined bands on chromosomes, so the location of genes is described according to the number of the chromosome on which they are found, whether they are on the long (q) or short (p) arm, and the band number. For example, the location of the α1-antitrypsin gene is described as 14q31–32.3, meaning that it is found on the long arm of chromosome 14 in the region of bands 31–32.3. The locations of some of the genes that have been mapped to the X chromosome are shown in Figure 43.1. FIGURE 43.1 The mapping of genes to specific sites on the X chromosome. The regions in the X chromosome where the genes associated with Duchenne and Becker muscular dystrophy (the dystrophin gene at p21.2), Lesch–Nyhan disease (the hypoxanthine-guanine phosphoribosyl transferase or HGPRT gene at q26.1-q26.2), fragile X syndrome (the FRAXA gene at q27.3) and glucose 6-phosphate dehydrogenase (the G6PD gene at q28) deficiency are located are shown. After the double helical structure of DNA had been discovered in 1953, the rather abstract concept of a gene became more tangibly associated with a physical structure. Nucleic acids consist of two complementary polymers of nucleotides. Each nucleotide consists of a purine or pyrimidine base, linked to a phosphorylated pentose. In DNA, the pentose is deoxyribose and the bases are adenine (A), guanine (G), cytosine (C) and thymine (T). In ribonucleic acid (RNA), the pentose is ribose and the pyrimidine uracil (U) replaces thymine. Protein coding sequences (exons) are interrupted by non-coding sequences (introns), which are variable in number (up to 50 in collagen genes, for example) and size (up to several thousand base pairs). As a consequence, although the knowledge that three bases code for an amino acid allows us to predict that the coding sequence for an average protein of 400 amino acids will be 1200 nucleotides, the complete gene could be an order of magnitude larger. The boundaries between exons and introns are critically dependent on the GT-AG rule: that is, introns almost invariably begin with GT (or GU in RNA) and end with AG. The structure of a hypothetical gene is shown in Figure 43.2. The promoter region of DNA, which precedes the coding region (‘upstream’ from the 5′ end of the gene, that is, in the opposite direction to transcription) is intimately involved in permitting and regulating expression. Some genes code for RNA (e.g. ribosomal and transfer RNA) that is not translated into protein, and modifications in the process of intron removal can result in one sequence of DNA participating in synthesis of different proteins, so that certain genes can be considered to overlap. FIGURE 43.2 The structure of a hypothetical gene. Coding sequences (exons) are shown in black, introns and non-coding regions are shaded and regulatory regions are in dark shading. Bases in exon sequences are shown in upper case, while bases in intron sequences are shown in lower case, illustrating the GT-AGT rule for starts and ends of introns. The Human Genome Project (HGP) represents an outstanding piece of multinational cooperation to map the entire human DNA sequence. The project, started in 1990, had several aims, the first of which was to determine the entire base pair sequence. The sequence of 3 billion base pairs was announced in draft form in 2000 and the complete sequence in 2003. It was thought initially that the human genome would consist of approximately 100 000 different coding genes. As the HGP neared completion, it emerged that the actual number would be closer to 30 000. Protein coding sequences and introns account for about 20% of DNA. The function of the remainder is being elucidated but in 2012, the initial findings of the ‘Encode’ project, which had been examining what had previously been called ‘junk’ DNA, were published. It appears that the remaining 80% does have a function within the genome, with much of the non-protein coding DNA appearing to code for RNA transcripts that may have other regulatory functions alongside those of gene enhancers and promoters. That the number of genes is much lower than expected appears to be because many genes can perform multiple functions, and it now appears that these functions may be regulated by the remainder of the genome. These discoveries have implications for the investigation and diagnosis of genetic disease, and issues concerned with gene expression will have a growing role in clinical genetics. The differentiated properties of each cell are determined by the pattern of genes in the cell that are active or dormant. In any one cell, only a small percentage of genes are likely to be actively engaged in directing synthesis of RNA at any one time. Many of these are ‘housekeeping’ genes, which are expressed in virtually all cell types. Much remains to be learned concerning the factors that determine whether or not a gene is expressed, but regulation of gene expression is clearly determined by proteins that interact with DNA. In higher (eukaryotic) organisms, DNA is found within the nucleus and the mitochondria, although the mitochondria have a very small percentage of the total DNA within the cell and a very small number of genes. Nuclear DNA is complexed with basic proteins (histones) to form chromatin. At regular intervals, DNA is wrapped around complexes of eight histones to form nucleosomes. The copying of DNA into RNA (transcription) is performed by RNA polymerase, which initiates transcription by interacting with the promoter region of a gene. Gene expression is inhibited if nucleosomes cover a promoter region and many factors that regulate transcription probably do so by competing with histones for binding to the promoter region. Within each promoter region, there are several elements that bind specific proteins capable of interacting with RNA polymerase and associated proteins. One of the key proteins in this group binds to the so-called ‘TATA box’ element (actually a TATAAAA or related sequence), which is found in most eukaryotic promoters and is usually located around 30 base pairs (bp) upstream from the transcription start site. Other conserved sequences, such as CAAT, also bind transcription factors and are found within the promoter region. Another class of regulatory sequence in DNA, the enhancer region, binds regulatory molecules, which include the steroid hormone receptors. Enhancer sequences may be some distance from the gene that they regulate, but the proteins that bind to them may nevertheless interact with the transcriptional apparatus as a result of looping in the DNA molecule. The initial RNA transcript (pre-mRNA) is modified in several ways before it leaves the nucleus (Fig. 43.3). First, a ‘cap’ structure (7-methylguanosine) is attached to the 5′ end and a sequence of about 200 adenylic residues (poly A) is added at the 3′ end. The non-coding introns are then removed by a two-step splicing mechanism to form a mature messenger RNA (mRNA). Splicing, which takes place within ‘spliceosomes’ (complexes of RNA and proteins), requires cleavage at the 5′ and 3′ ends of the intron and ligation (joining) of the exons. The specific boundary sequences found at splice junctions (see above) act as signals for splicing. Comparison of large numbers of splice junctions reveals these ‘consensus sequences’ to be of the general form AGgta at the 5′ junction and cagG at the 3′ junction (where bases in the intron sequence are in lower case). Given that pre-mRNA molecules can contain up to 65 exons and an intron may consist of thousands of nucleotides, it is remarkable that the correct sites for splicing can be chosen. FIGURE 43.3 Transcription and mRNA processing. After transcription, processing of precursor mRNA involves capping, whereby GTP is attached to the 5′ end of the mRNA precursors via a 5′-5′ triphosphate linkage (i.e. in the reverse orientation to all other nucleotides); addition of around 200 adenylate residues to form a poly(A) tail at the 3′ end; and splicing, in which introns are excised and exons spliced together. Translation of mRNA into protein is initiated by ribosomes and transfer RNA at the AUG codon and terminated at one of the stop codons (UAG, UAA or UGA). Finally, the process of translation involves the activity of ribosomes, transfer RNA and a variety of other molecules which synthesize a protein using the mRNA code as a template. A group of three nucleotides (a codon) specifies an amino acid and most amino acids are coded for by more than one codon (i.e. the genetic code is degenerate). In principle, each RNA sequence can be decoded in three different reading frames, depending on which triplet is chosen as the first codon. In practice, the reading frame is determined by the site of initiation, which always occurs at an AUG codon (AUG codes for methionine, but the initiating methionine is cleaved from proteins in eukaryotic cells). Translation stops at any one of three stop codons (UAA, UAG or UGA). Any subsequent modification of a protein, such as proteolytic cleavage or addition of carbohydrate, is known as post-translational modification. The accepted terminology, which referred to the ‘normal’ gene in a population as the ‘wild type’, has changed and the terms ‘normal’ and ‘mutant’ (or ‘variant’ if pathogenicity is unclear or questionable) are now preferred. However, the genetic constitution of populations is in a constant state of flux with new genes appearing as a result of mutation and deleterious genes being removed by natural selection. Mutations can be broadly divided into those that change the genetic code at a specific location (point mutations or single nucleotide polymorphisms, SNPs) and those that result in gain or loss of genetic material (deletions, duplications and insertions). Point mutations can result from incorrect insertion of a base during DNA replication by DNA polymerase or spontaneous decomposition reactions such as depurination and deamination. Mutagenic chemicals that increase this error rate include those that mimic the natural bases or distort the structure of DNA and those that chemically modify DNA. Ultraviolet light also causes point mutations, particularly by formation of pyrimidine dimers. Point mutations in which a purine is replaced by another purine (e.g. A replaced by G) or a pyrimidine is replaced by another pyrimidine are known as transitions, while replacement of a purine by pyrimidine (e.g. G replaced by C) or vice versa is known as a transversion. Gain or loss of genetic material can result from various errors, including chromosomal breakage and unequal crossing over. Insertion of viral sequences into DNA can also disrupt the genetic code and the rate of spontaneous chromosomal breakage can be markedly increased by ionizing radiation. Whenever the number of bases deleted or inserted is not a multiple of three, the reading frame of the mRNA is altered (frameshift mutation) and the RNA sequence subsequent to the mutation becomes nonsense. Accumulated damage to DNA would rapidly overwhelm the organism, but repair mechanisms recognize and repair damaged DNA so that fewer than 1 in every 1000 accidental base changes results in a stable mutation. It is estimated that stable point mutations are acquired at a rate of about 1 in every 109 base pairs during each cell generation. Consequently, an average gene of about 103 coding base pairs is likely to acquire a mutation once in every 106 cell generations. As might be expected, individuals with inherited defects in the enzymes responsible for DNA repair are markedly more susceptible to the effects of environmental mutagens. A significant proportion of germline point mutations are thought to be caused by modification of methylated cytosine residues. DNA methylation, restricted in eukaryotic cells to cytosine residues, usually at CpG dinucleotides (CpG denotes C-phosphate-G in a linear sequence, in distinction from a CG base pair) is not present in all such organisms but is thought to play an important role in ensuring stable inheritance of expression patterns when cells divide. Spontaneous deamination of the 5-methylcytosine creates thymidine, and problems can then arise when the normal guanine on the complementary strand becomes an adenine and the mutation cannot be detected by DNA repair mechanisms. Consequently, methylation of cytosine can produce mutation ‘hot spots’ (i.e. sequences associated with an unusually high frequency of mutations or recombination). Some mutations may have no effect on the structure of a protein – either because the genetic code is degenerate and the new sequence codes for the same amino acid, or because some amino acids in a protein can be substituted without producing any significant effect on the function of the protein. However, some apparently ‘silent’ mutations may have an effect on the protein product, not because of the actual base change involved, but rather through an effect on splicing by activating cryptic splice sites or destroying splice enhancers. Mutations that change the three-dimensional structure of a protein and so alter its function or stability may do so by a variety of mechanisms (Fig. 43.4). FIGURE 43.4 Examples of mutations. (A) G to A transition in the α1-antitrypsin gene, a missense mutation resulting in substitution of glutamic acid by lysine at position 342, producing the Z variant associated with α1-antitrypsin deficiency. (B) Transversion of C to T in the steroid 21-hydroxylase gene, converting the codon for glutamine to a stop codon, one of the mutations causing congenital adrenal hyperplasia. (C) G to A transition in the 5′ splice site of intron 12 in the phenylalanine hydroxylase gene, resulting in deletion of exon 12, the most frequent cause of phenylketonuria in Caucasians. Bases in intron sequences are shown in lower case. (D) Deletion of three bases in the cystic fibrosis transmembrane conductance regulator (CFTR) gene resulting in deletion of phenylalanine at position 508, the most frequent mutation causing cystic fibrosis in Caucasians. (E) Insertion of a G in the hypoxanthine-guanine phosphoribosyl transferase gene, a frameshift mutation that results in the Lesch-Nyhan syndrome. (F) Amplification of a CGG triplet repeat in the FMR-1 gene, which causes the fragile X syndrome. By convention, the DNA strand that has the same sequence as mRNA (except that it possesses T instead of U) is represented. This strand is known as the coding strand, although it is the ‘anticoding’ strand that is complementary to mRNA and therefore provides the template for mRNA synthesis. Some amino acid changes (missense mutations), such as that which produces the Z variant of α1-antitrypsin (Fig. 43.4A), can have a profound effect on the processing or function of a protein. Some mutations (nonsense mutations) create or destroy codons for the start or stop signals of translation so that a protein of abnormal length is produced (Fig. 43.4B). Mutations at splice sites (Fig. 43.4C) frequently result in production of abnormal mRNA that is unstable. Deletion of three bases removes the codon for a single amino acid without altering the reading frame, as occurs in the most common mutation causing cystic fibrosis (Fig. 43.4D). Insertion (Fig. 43.4E) or deletion of any number of bases that is not a multiple of three will alter the reading frame so that the message becomes garbled. Amplification of triplet repeat sequences (Fig. 43.4F) has been identified as the basis of several inherited diseases. Occasionally, mutations affect regulatory regions of DNA so that the amount of protein produced is altered. Although mutations are most frequently either neutral or deleterious, rare mutations will alter the function of a protein in such a way that the fitness of an individual is improved, so contributing to evolution. Inheritance of the mutations that have accumulated in our ancestors, whether they be advantageous, neutral or deleterious, is what constitutes our individuality. An individual inherits two copies of each chromosome (one maternal and one paternal). On each chromosome, the sequences at each site, or locus, are known as alleles. If the two alleles are identical, the individual is said to be homozygous at that locus, while if the alleles are different, the individual is said to be heterozygous for each allele. As will be seen later, genetic disease is usually heterogeneous, so that an individual said to be homozygous for a deleterious gene may be found, when studied at the molecular level, to carry a different mutation in each allele (i.e. is a compound heterozygote). When the prevalence of a mutant allele becomes more common in a population than could be maintained by new mutations alone (generally taken to be when more than 1% of the population carry the allele), there is said to be polymorphism. Many proteins in blood (e.g. haptoglobin) and on cell surfaces (e.g. human leukocyte antigen, HLA) are polymorphic. The classic inherited diseases result from single gene defects and more than 6000 inherited diseases likely to be associated with defects in single genes have already been identified. Inherited diseases could, in theory, result from mutations in any one of the human genes – the only limitation being that the structure of some gene products is so critical that any mutation will not be compatible with life. Most of the more common diseases that afflict western society, including most cases of diabetes, atherosclerosis and hypertension, are the result of interaction between the environment and polygenic factors (i.e. they are determined by interactions between several genes). Molecular analysis of the polygenic diseases is considerably more difficult than analysis of single gene defects, but alleles that predispose individuals to development of these diseases are being identified. Both single gene defects and most multifactorial/polygenic diseases arise from mutations in the nuclear DNA but genetic diseases also arise from mutations in mitochondrial DNA and from chromosomal abnormalities. Mitochondrial DNA is extranuclear and shows almost complete maternal inheritance. Chromosomally inherited disorders include the trisomies, where faulty meiosis allows two copies of a chromosome to be present in a gamete, leading to three copies in the embryo. Trisomy of chromosome 21, for example, is responsible for Down syndrome. If new mutations occur in germ cells, then they may give rise to an inherited disease in the next generation. The effects of mutations on non-germ cells, or somatic cells, will depend both on the gene affected and on the state of differentiation of the cell affected. The ageing process is likely to be one result of accumulated mutations in somatic cells and the central role of mutations for the development of cancer has become clearer during the last few years. An individual’s genotype is determined at the time of fertilization, when the chromosomes of the gametes (i.e. the male sperm and female egg) are combined. The formation of gametes (gametogenesis) is particularly relevant to an understanding of the detection of inherited disease because it is at this stage that ‘shuffling’ of genes occurs. Normal cell division, or mitosis, involves a simple copying of each chromosome, with one identical copy being passed on to each daughter cell. To avoid a doubling in the number of chromosomes in each generation, gametogenesis involves a reduction (by half) of the chromosome number during two specialized cell divisions known as meiosis. Since the chromosome complement of parent and offspring must be equivalent, the reduction in chromosome number cannot be arbitrary: parental contributions must be equal and equivalent. This requirement can be met because each somatic cell of an individual is diploid, containing corresponding (homologous) pairs of chromosomes, one derived from the mother and the other from the father. Meiosis consists of two cell divisions: in the first, after duplication of DNA, pairing of homologous chromosomes occurs, and to ensure that each gamete receives just one member of each homologous pair, the duplicated paternal chromosome is distributed to one, the duplicated maternal chromosome to the other. The assortment between the two cells appears to be random so that each cell acquires some maternal and some paternal chromosomes. The second cell division is like ordinary mitosis, except that it is not preceded by duplication of chromosomes. As a consequence, the gametes produced are haploid, with half the normal number of chromosomes. At first sight it might be expected that a chromosome would be transmitted from one generation to the next as an intact unit and that two genes on the same chromosome would always be inherited together. The fact that this is not so is a result of events that occur during the first meiotic division, which have important consequences for genetic analysis. As chromosomes pair prior to the first division, crossovers (or chiasmata) occur by breakage and rejoining between the chromatids of homologous chromosomes resulting in recombination (Fig. 43.5). The recombination fraction is a measure of the genetic, rather than the physical, distance between two genes (or loci). The recombination fraction for two loci can never be more than 0.5 as the resulting chromatids can only ever be recombinant or non-recombinant, no matter how many crossovers have occurred between the loci. In simple terms, linkage of two genes (i.e. a tendency to be inherited together) occurs only when the genetic distance separating them is sufficiently short to make crossover between them unlikely. The association of two genes on separate chromosomes is random but association of genes on the same chromosome is not, as it is known that crossovers do not occur at random. A process known as interference prevents a chiasma forming if one already exists nearby. When genes are associated more frequently than would be predicted by chance, they are said to be in linkage disequilibrium. FIGURE 43.5 Crossover and linkage. Exchange of alleles as a result of crossover between homologous chromosomes. Linkage between alleles a and b (or A and B) occurs if they are sufficiently close that crossover is unlikely to occur between them. In females, gametogenesis is initiated during fetal development and, at birth, the germ cells are in a phase of arrested maturation of the first stage of meiosis, which is not completed until ovulation. The increased risk of chromosomal abnormalities in older mothers may be explained by the fact that completion of meiosis occurs only after ovulation, when the second stage of meiosis (which is similar to ordinary mitotic division) occurs and during which fertilization can take place. This may happen up to 50 years after formation of the germ cells. In males, sperm production continues from the time of sexual maturity into old age and the large number of cell divisions involved is probably the cause for the increased number of new single gene mutations that seem to occur in the children of older men. Mendel introduced the concepts of dominant ‘characters’ (or traits), which are transmitted without change, and recessive traits, which become latent after cross-fertilization. In modern terms, an allele coding for a dominant trait can be said to manifest its phenotype in the heterozygous state (i.e. only one copy is required for its effects to be apparent), while an allele for a recessive trait expresses its phenotype only in homozygotes. An individual who is heterozygous for an autosomal recessive condition is described as a carrier. The types of family pedigree associated with autosomal dominant and recessive genes are illustrated in Figure 43.6A and B. Autosomal dominant conditions affect males and females equally, and affected individuals who are heterozygous for the abnormal allele will transmit it to half of their offspring. Autosomal recessive disorders occur in individuals whose parents are both carriers for the mutant gene. The risk for such patients of having affected children is 25% and the probability of any child they have being a carrier is 50%. Dominant diseases are often associated with genes coding for proteins that have structural, carrier or receptor functions, while genes coding for enzymes are often associated with recessive disorders. The explanation for this is probably that the activity of most enzymes is considerably greater than necessary for normal metabolism, so loss of up to half of the normal activity is of little consequence. Inheritance is somewhat different for alleles on X chromosomes. Dominant X-linked diseases will affect both males and females, but an X-linked recessive disease will be manifest only in males, who have only one X chromosome (Fig. 43.6C). In females, during the very early stages of embryogenesis, one or other X chromosome is inactivated in every cell; consequently, females can be carriers but will usually only suffer from an X-linked recessive disease if they are homozygous. However, sometimes, in some disorders, the inactivation is not random but occurs in such a way that only the normal chromosome is inactivated, with the result that only mutant alleles are expressed in critical tissues (‘skewed’ X-inactivation) and the individual becomes a manifesting heterozygote. This has been reported in several X-linked recessive disorders including, for example, Duchenne muscular dystrophy. FIGURE 43.6 Patterns of inheritance in families. Inheritance of (A) a dominant condition, (B) a recessive condition and (C) an X-linked recessive condition. Squares represent males and circles represent females. Open symbols represent normal individuals, while fully shaded symbols refer to affected individuals and half-shaded symbols represent carriers. When both alleles are expressed in a heterozygote, each producing its phenotype independently, inheritance is said to be codominant. This type of inheritance is seen most clearly when phenotypes are determined by immunological or biochemical tests – for example, in testing for blood groups and in restriction fragment length polymorphism (RFLP) analysis and, more recently, in the growing field of pharmacogenetics (see later), where it is possible to distinguish both alleles at each locus. Of the inherited diseases currently recognized, the overwhelming majority are due to problems in nuclear DNA and are autosomal dominant, autosomal recessive or sex linked. However, it is now known that some diseases are associated with the small amount of DNA that is present in mitochondria. These diseases are maternally inherited, since the mitochondria in the fertilized egg are of maternal origin. The relative frequency of inherited disease varies markedly between populations: for example, cystic fibrosis and α1-antitrypsin deficiency are associated primarily with northern Europeans, while red cell disorders (thalassaemia, sickle cell anaemia and glucose 6-phosphate dehydrogenase deficiency) are found primarily in people of Mediterranean, Oriental or African origin, and Tay–Sachs disease is found primarily in Ashkenazi Jews. In some populations, the prevalence of an inherited disease is due to a ‘founder effect’, as with variegate porphyria in South Africans, which can be traced to a single couple who emigrated from Holland in the 1680s. Autosomal recessive diseases that are particularly widespread in larger populations are likely to represent a balanced polymorphism in which disadvantage to homozygotes is balanced by an advantage to the larger number of heterozygotes. With some red cell disorders (e.g. HbS, the sickle cell trait), the balanced polymorphism is clearly a response to the environment, in this case with heterozygosity conferring resistance to malaria caused by Plasmodium falciparum. In a large population, the relative frequencies of different alleles tend to remain constant and a simple mathematical formula allows calculation of the frequency of different genotypes. If two alleles, A and a, occur at a given locus and their frequencies are p and q, respectively, then: It can be shown that the genotypes AA, Aa and aa have frequencies p2, 2pq and q2, respectively (the Hardy–Weinberg law). Use of this law allows simple calculation of carrier frequencies for autosomal traits. For example, if the homozygote frequency (q2) for cystic fibrosis is about 1 in 2500, q is 1/50, p is 1 − q or 49/50 (~ 1) and the heterozygote frequency (2pq) is about 1/25. Several factors dictate that each genetic disease is associated with symptoms of quite variable nature and severity. It is often naively assumed that all mutations of a particular gene will have identical consequences for the organism, but this is far from being true. Different mutations in a given gene are quite likely to give rise to different phenotypes, just as a particular phenotype can result from many different mutations in the same gene or even in different genes. Since many different mutations that have deleterious effects on a particular gene are present in most populations, it is not surprising that molecular analysis often reveals that individuals who have been described as homozygous are in fact compound heterozygotes, that is, they are affected by two different deleterious alleles. Because the haemoglobin gene has been investigated in detail, it provides a good illustration of the complexity of genetic disease. Several hundred abnormal haemoglobins have been identified. The majority have amino acid substitutions resulting from single base changes and the consequences range from complete absence of protein to variant haemoglobins with function indistinguishable from that of normal (see Chapter 29). As study of other genes progresses, similar complex arrays of mutations of every imaginable type and with differing consequences are being discovered. With the introduction of screening for phenylketonuria (PKU), it soon became apparent that the disease is heterogeneous. It has been shown that, in PKU, phenotypic heterogeneity is related to the level of phenylalanine hydroxylase activity expressed in each patient which, in turn, is determined, in classic PKU, by the particular mutations that are present in the two alleles of the phenylalanine hydroxylase (PAH) gene. However, not all cases of neonatal hyperphenyalaninaemia are due to PAH deficiency. Benign hyperphenyalaninaemia arises from a transient liver immaturity and does not lead to the disease. Two rare causes of PKU are deficiencies in the enzymes dihydropteridine reductase and dihydrobiopterin synthetase, leading to PKU with a severe phenotype. Glucose 6-phosphate dehydrogenase (G6PD) deficiency, which is an X-linked recessive condition estimated to affect as many as 500 million people worldwide, has also proved to be extremely heterogeneous at both the phenotypic and molecular levels. Some variants appear to have no clinical consequences, while at the other extreme, severe defects in G6PD cause hereditary non-spherocytic haemolytic anaemia (see Chapter 27). Intermediate defects are associated with haemolytic anaemia only in the presence of precipitating factors (e.g. infection, ingestion of fava beans or certain drugs). Analysis of the protein had indicated the existence of about 400 variants of G6PD and a similar number of mutations have now been described, but many of these appear not to cause disease. Many of the mutations causing the most severe disease are clustered near the carboxy end of the enzyme in the region of the putative NADP binding site. A further level of heterogeneity in genetic disease occurs as a result of varying degrees of penetrance and expressivity. Penetrance refers to the degree to which the mutation causes disease. Thus, in some disorders, the presence of the mutant gene is disease-causing in some individuals but not others, demonstrating variable penetrance, whereas in diseases that are fully penetrant, the presence of the mutation will always lead to the disease phenotype. For example, the C282Y (Cys282Tyr) mutation in the HFE gene (causing substitution of the cysteine residue at position 282 of the human haemochromatosis protein by tyrosine), causes haemochromatosis in some individuals but results in a completely normal phenotype in others. Expressivity is a slightly different aspect of a gene’s effect and refers to the presence of variable phenotypes arising from the same mutation. It can be age-related or determined by the environment (as when drugs such as barbiturates precipitate attacks of acute intermittent porphyria). An example of a disorder demonstrating variable expressivity is Waardenburg syndrome, where the ‘full’ syndrome includes several phenotypic features (such as different coloured eyes, a white forelock or deafness) but where, within a single affected family, different individuals may have only one feature, which is not always the same one within that family. Variable expressivity and penetrance tend to be features of dominant, rather than recessive, conditions. Another aspect of inheritance, known as imprinting, may help to explain processes such as variable penetrance and expressivity. Contrary to the assumptions of classic genetics, it now appears that expression of some genes depends on whether they are of maternal or paternal origin. The molecular mechanism of imprinting, which is likely to occur during meiosis, involves DNA methylation, which ‘marks’ certain genes and ensures that they are preferentially expressed in the next generation. If such a gene is imprinted in the maternal line, it will continue through a woman’s daughters but not through her sons, although both may be affected. The converse is true for paternally imprinted genes, which will be transmitted through sons but not through daughters. Consequently, if an imprinted gene contains a deletion, offspring will not show expression of the gene product even in the presence of a normal gene on the opposite chromosome, as this will be ‘switched off’. Prader–Willi and Angelman syndromes are good examples of disorders arising due to abnormalities in a region carrying imprinted genes (chromosome 15 at 15q12,). Prader–Willi syndrome, features of which include hypotonia and hyperphagia, is produced by deletion of the paternal alleles at 15q12. Angelman syndrome, which is associated with ataxic movements and seizures, is also associated with deletion of 15q12 but, in this case, of the maternal allele. In some cases of Prader–Willi syndrome, rather than deletion of the paternal alleles, loss of the paternal chromosome occurs together with maternal isodisomy (two copies of the same allele from the mother) or heterodisomy (one copy of each maternal allele). There are several possible mechanisms by which two alleles can be inherited from one parent. Trisomies, for instance, usually result in spontaneous abortion, but if one chromosome is then lost there is a one in three chance that resulting cells will have a normal complement of chromosomes but with one pair of chromosomes derived from a single parent. The interesting possibility that a recessive disease can be inherited from one carrier parent then arises. This unusual mode of inheritance has been demonstrated in some patients with cystic fibrosis, but it is not yet clear how frequently it occurs in this or other diseases. Analysis of DNA is heavily dependent on the availability of techniques to identify specific nucleotide sequences. Fortunately, the function of DNA has resulted in the evolution of proteins capable of recognizing specific DNA sequences and it is inherent in the structure of DNA that one strand should recognize and bind (hybridize) specifically to its complementary strand. Most of the techniques currently used in DNA technology exploit one or other of these properties. Without restriction enzymes, much of the molecular biological analysis carried out in the last 30 years would not have been possible. These enzymes are widespread in bacteria: over 3000 having been recognized so far, of which around 600 are available for commercial/analytical use. Each enzyme is named after the species of bacteria in which it was found (e.g. EcoRI from Escherichia coli), and in which it probably fulfils a defensive function, cleaving molecules of foreign DNA. The usefulness of these enzymes derives from the fact that they do not cleave DNA at random, but recognize and cut specific nucleotide sequences. The most commonly used restriction enzymes recognize sequences of 4–6 nucleotides that have a two-fold axis of symmetry and are therefore said to be palindromes (i.e. the sequence reads the same on the complementary strand) (Fig. 43.7A). Digestion of DNA by a particular enzyme provides reproducible fragments whose size will depend on the frequency with which the enzyme recognition site occurs. On average, a 4-bp site occurs every 256 bp and a 6-bp site every 4096 bp. While some enzymes (e.g. HaeIII) cut in such a way that ‘blunt ends’ are produced, others (e.g. EcoRI) cut asymmetrically so that ‘sticky ends’ are left, which are extremely useful for reannealing fragments to produce recombinant DNA (Fig. 43.7B). FIGURE 43.7 Restriction enzymes and recombination. (A) Arrows indicate how specific DNA sequences are cleaved by restriction enzymes. (B) Cohesive ends produced by restriction enzymes can be used for annealing of DNA sequences, which can then be joined by a ligase to form a recombinant DNA molecule. In addition to allowing reproducible cleavage of DNA to a manageable size, restriction enzymes are also valuable tools for analysing molecular diversity and identifying the individuality of DNA sequences. Differences in DNA sequences between individuals may create or destroy sites for restriction enzymes (i.e. there is polymorphism of restriction sites). Thus, the distance between restriction sites will often differ between individuals and between the maternal and paternal strands of DNA. The pattern of restriction sites can therefore provide a ‘signature’ for each individual strand of DNA. The different populations of DNA fragments produced on digestion by an enzyme are known as restriction fragment length polymorphisms or RFLPs. Restriction enzymes have more recently acquired a new use in preparing genomic DNA for the various techniques employed in ‘next generation’ sequencing. A probe is a sequence of DNA (or RNA) that has been labelled in order to identify complementary base sequences by molecular hybridization. The two strands of DNA can be dissociated (‘denatured’ or ‘melted’) in various ways, such as by heating or addition of alkali. Denaturation for a given fragment of DNA occurs at a specific temperature, and the temperature at which 50% of the duplex is dissociated is known as the Tm. When the temperature is lowered to just below the Tm, hydrogen bonds begin to reform between complementary bases, a process known as annealing or renaturation. If the probe and target DNA are mixed before reannealing is allowed to occur, the probe can be used to ‘find’ its complementary sequence. The conditions under which reannealing occurs (in particular salt concentration and temperature) determine the degree of stringency of the hybridization. Probes can be used to detect their complementary sequences, traditionally after DNA (or RNA) fragments have been separated by electrophoresis. Digestion of genomic DNA with a restriction enzyme will produce a million or so fragments of different sizes and electrophoresis has the great advantage of allowing simple determination of the size of fragments detected by probes. The now classic technique of Southern blotting involves the transfer of electrophoretically separated bands of DNA to a sheet of nitrocellulose or nylon. Complementary DNA sequences are then detected by hybridization with labelled probes. The technique was originated by Dr (later Professor) E M Southern. Subsequently, the terms Northern and Western blotting have been used for processes in which RNA or proteins, respectively, are transferred. In the past, all DNA analysis required some technique of visualizing the products of the reaction, and with probe hybridization this involves labelling the probe in some way. 32Phosphorous radioactive labelling, although still in use for blotting protocols, was superseded by the use of fluorescent dyes, particularly for gene sequencing, a system which allowed the development of large, high-throughput, DNA analysers, as well as techniques such as real-time PCR. However, some of the technologies employed in ‘next generation’ sequencing systems require neither gels nor dyes, as will be discussed later. Some of the newer systems allow for an electrical signal to be generated when hybridization of probe to target occurs, which may eventually lead to the development of point-of-care systems. Probes, generally of a few thousand bases, may be sequences cut from genomic DNA or they may have been produced by making complementary DNA (cDNA) to an mRNA species. The latter procedure uses the enzyme reverse transcriptase, which transmits genetic information in the ‘reverse’ direction, that is, from RNA to DNA. Genomic and cDNA sequences differ, particularly in the absence of intron sequences from the latter. The tolerance of probes for mismatching of base sequences will depend on their size and on the stringency of hybridization. Shorter oligonucleotide probes are often more useful for direct identification of point mutations since conditions can be chosen such that hybridization occurs only when there is complete complementarity between probe and target. Because hybridization of a relatively smaller proportion of bases in the probe is required, larger genomic or cDNA probes will usually recognize corresponding sequences of DNA from different individuals in a population or even different but related genes. Cloning, where the sequence of interest was ‘grown’ in bacteria such as E. coli after being inserted into the bacterial genome using bacteriophage viruses, has been largely superseded as a method of producing probes by the ability to create synthetic oligonucleotide sequences, although cloning as a technique is still employed in areas of research. Without doubt, the commonest use for synthetic oligonucleotides has been as ‘primers’ in the polymerase chain reaction (PCR). After its introduction in 1985, PCR supplanted many of the more tedious techniques of molecular biology and opened up completely new possibilities. Essentially, a means of cloning DNA without the need for vectors or bacteria, PCR uses the enzyme DNA polymerase to copy DNA. To do this, the enzyme needs two oligonucleotide primers that are complementary to sequences flanking the region of interest in the target DNA, with one on each strand (Fig. 43.8). Computer and web-based programs are available for designing primers (usually 20 or more bases) in order to choose sequences likely to be most suitable for the PCR reaction and to maximize specificity by ensuring that the complementary sequence is virtually unique in the genome. The target sequence is amplified exponentially by repeated cycles of enzymatic copying. In the first cycle, double-stranded DNA is denatured by heating to between 92°C and 96°C for 5 min, and then cooled to (usually) between 55°C and 60°C, so that the oligonucleotide primers can anneal to their complementary sequences in the target DNA. For optimal specificity, the highest annealing temperature possible is used to minimize extension of primers bound non-specifically. Extension of the primers by DNA polymerase, using added nucleotides and the DNA target as a template, is then allowed to take place. Use of a heat-stable DNA polymerase, which can withstand the heating cycles, avoids the necessity of adding fresh enzyme at each cycle and allows extension to take place at a high temperature (72°C). After heat denaturation, excess primers can then anneal to the newly synthesized DNA as well as the original DNA strands and the process is repeated. In 30 cycles, amplification of over one million-fold can be achieved. Originally, the only enzyme available for PCR was that from Thermus aquaticus (Taq polymerase), but more have now been identified and developed, making amplification of much longer sections of genomic DNA possible. FIGURE 43.8 The polymerase chain reaction. After denaturation of DNA, primers anneal to complementary sequences. During the first cycle, a heat-stable DNA polymerase (usually Taq) initiates synthesis of two new strands. After a further denaturation cycle, primers anneal to the newly synthesized DNA as well as the original sequences and four new strands are synthesized (cycle 2). With an exponential increase in the number of DNA strands, after around 30 cycles the sequence will have been amplified about one million times. Stringent control of conditions, such as the concentrations of magnesium and nucleotides in the reaction mixture, have usually been needed to maintain the specificity of a PCR although conditions that maximize polymerase fidelity may reduce PCR efficiency. Where such manipulation was necessary, it was predominantly dependent on the sequences involved, especially that of the primers. However, the ability to amplify longer sections of DNA means that primers can be ‘picked’ to suit the conditions required, allowing the use of universal ‘master mixes’ and thus a greater standardization of PCR assays. PCR is so sensitive that it has been used to amplify the DNA from a single cell and it can be used with samples obtained from materials as diverse as ancient mummies, fossils, hair follicles, preimplantation embryos and fixed pathological specimens. The large quantities of amplified DNA produced by PCR, which can be detected using a variety of visual labels, have eliminated the need for radiolabelling. The extreme sensitivity of the technique is also the source of one of its disadvantages – contamination by extraneous DNA (e.g. from the operator) can create havoc, so that strict precautions must be taken to avoid artefacts. Non-isotopic fluorescent labels are employed in the widely used technique of real-time PCR. There are several versions of this technique but the basic principle is that a PCR reaction is followed, in real time, by monitoring the signals produced by dye-labelled probes that bind to the accumulating products. Various modifications to the technique allow real-time PCR to be used for quantitation of a target sequence (hence, its other, abbreviated name of qPCR) and determining dosage (the number of copies of a gene in a cell) and gene duplication as well as for mutation detection. Quantitative analysis has become increasingly important as a tool for microbiologists for determining such things as viral load in samples from patients suffering from diseases such as human immunodeficiency virus infection. Not to be confused with real-time PCR, reverse transcriptase PCR (rtPCR) uses purified RNA as the start point and uses the enzyme reverse transcriptase to produce cDNA, which can then be amplified using conventional PCR and used to examine the protein expressed by the gene of interest. Potentially, this allows investigation of both the effect of any sequence variants in a gene on its protein product, and the possible phenotypic consequences.
Molecular clinical biochemistry
INTRODUCTION
GENES AND GENE EXPRESSION
What is a gene?
The Human Genome Project
The ‘Encode’ project
Gene expression
Mutation, the source of diversity and disease
Genesis of an individual: the formation of gametes
Genes in families and populations
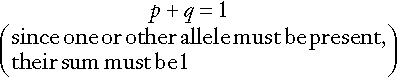
The variable expression of genetic disease
THE TECHNIQUES OF GENETIC ANALYSIS
Detection of specific sequences in DNA
Use of proteins that recognize DNA sequences: restriction endonucleases
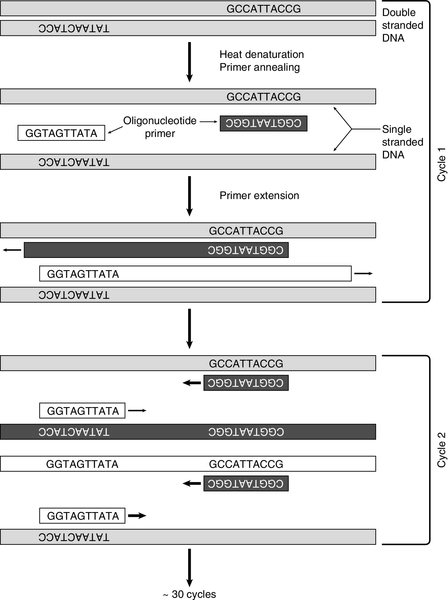
Hybridization: probes and the polymerase chain reaction (PCR)
Detection of mutations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


