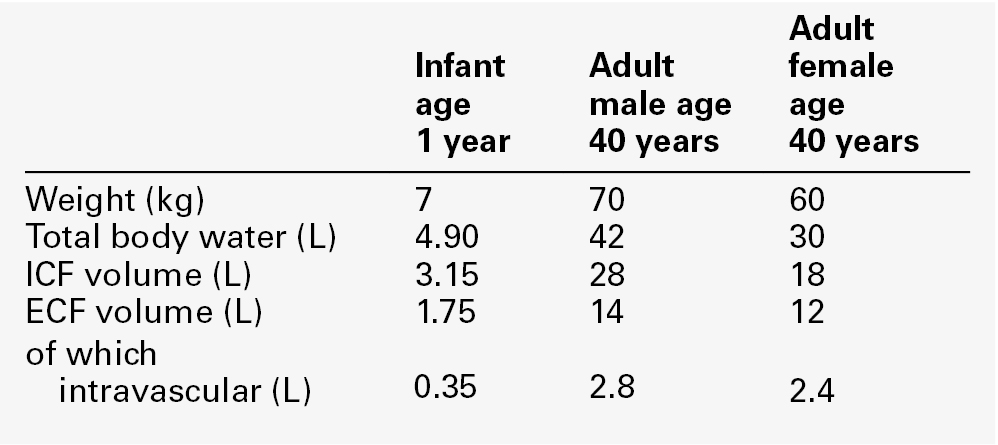
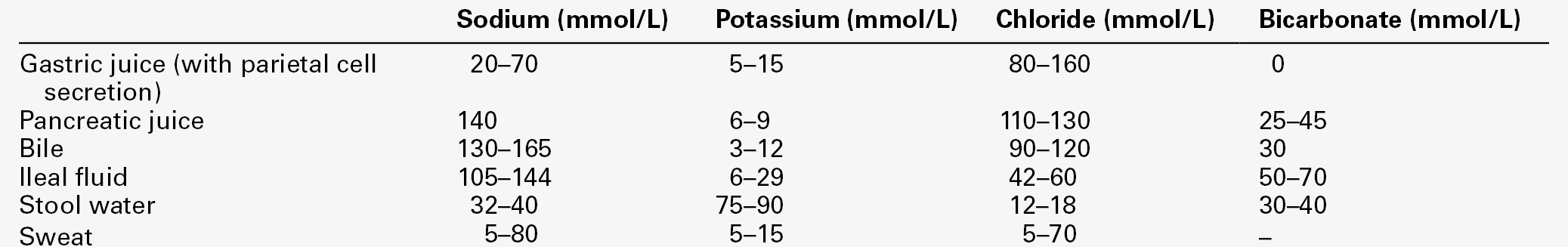
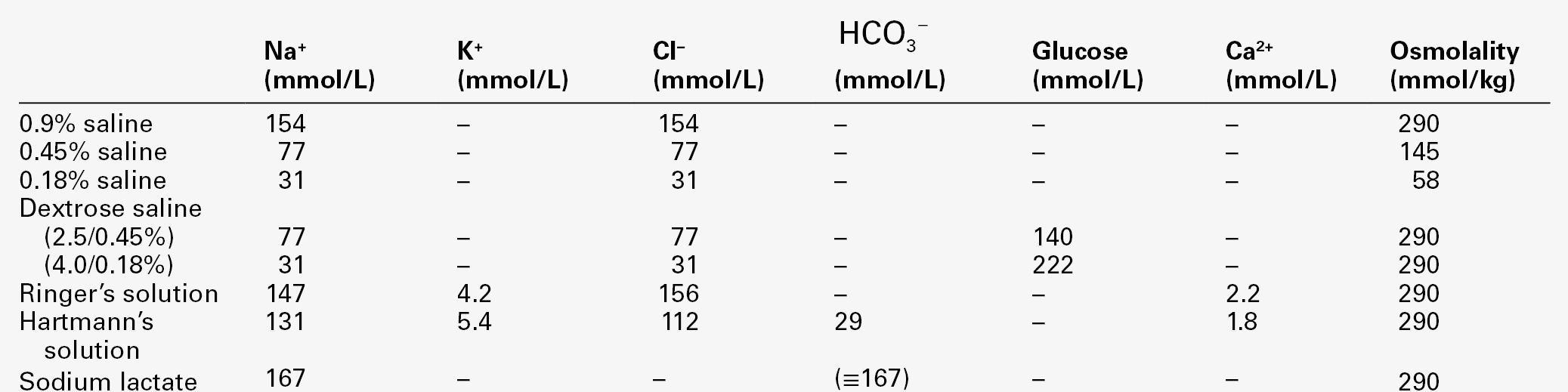
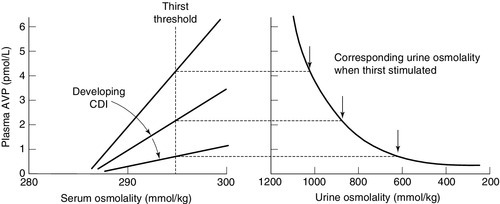
CHAPTER 4 CHAPTER OUTLINE Extracellular fluid and sodium Extracellular fluid, intracellular fluid and potassium DISORDERS OF SODIUM METABOLISM DISORDERS OF POTASSIUM METABOLISM Water is the most abundant molecule in the human body and is the major solvent (the only other of consequence is fat). The physiological control of the composition and distribution of the fluid spaces is a highly sensitive and complex homoeostatic process necessary to maintain a constant milieu interieur. Two main fluid spaces exist: the intracellular fluid (ICF) and the extracellular fluid (ECF). The latter is further separated into the intravascular space (plasma), the interstitial space (which includes lymph) and transcellular fluid (pleural, pericardial, peritoneal, cerebrospinal and gastrointestinal fluids), formed by the transport activity of cells. Table 4.1 summarizes the water content of the body and the distribution of fluid between the main body spaces; the proportion of body water to total body weight is affected by age, gender and body fat content. The electrolyte compositions of the ECF and the ICF are different – in essence, the extracellular space is a predominantly sodium-containing solution and the intracellular space a potassium-containing solution (Table 4.2). This fundamental difference in electrolyte composition is maintained by cell membrane transport pumps (energy-consuming ATPases). Electrolyte and protein concentrations of blood are now most commonly measured in serum. In this chapter, unless specified, the term plasma is used for describing in vivo concentrations and the term serum for measured in vitro concentrations. TABLE 4.2 Representative molal concentration of electrolytes within the body fluid spaces Body water moves between the main body spaces through water channels (aquaporins), predominantly under the influence of the osmotic pressures resulting from dissolved particles in the ECF and ICF on either side of the cell membrane. Under steady-state physiological conditions, the osmotic pressure of the ICF equates exactly with the plasma osmotic pressure. Osmolality represents the molal concentration of solute in a litre of solvent (water) and is expressed as mmol/kg, as opposed to a molar (or calculated osmolar) solution, which is the concentration in the space of a litre of solution (which includes solute space) and is expressed as mmol/L. This apparent nicety in definition does have its uses in differentiating certain real from apparent electrolyte disorders (see p. 46). The measured osmolality in ECF cannot, however, always be equated with transcellular osmotic pressure. The cell membrane is selectively permeable to a variety of solutes, but certain natural solutes such as urea, or exogenous solutes such as alcohol, are freely permeable. Thus, an increase in plasma osmolality due to sodium implies an increase in osmotic pressure across the cell membrane, tending to withdraw water from the cell to equalize osmolalities. However, an increase in plasma osmolality due to urea does not have this effect because of the free permeability of urea between the ICF and ECF. This leads to the concept of effective osmolality or tonicity, which, under physiological conditions, is primarily dependent on plasma sodium concentration, but under pathological or iatrogenic conditions, may be dependent on other solutes, for example, the effect of glucose in untreated diabetes mellitus or mannitol following its intravenous infusion (used therapeutically precisely for this effect). The distribution of water between different body spaces is thus dependent on the permeability of the relevant membrane barriers to water, and the quantity of solute within each space. Because water is freely permeable across all cell membranes (except some highly specialized membranes in the nephrons and sweat glands), the water content of body spaces is dependent on the solute content of the space. The Gibbs–Donnan effect is an important force that influences the distribution of solutes. If the barrier separating two compartments is permeable to water and small ions, but impermeable to large ionized molecules, and the larger molecules are confined to one compartment, the concentration of small ions will differ between compartments at equilibrium, and the compartment containing the protein will exert an osmotic force. Oncotic pressure (colloid osmotic pressure) is the osmotic pressure resulting from the difference within the ECF between the protein contents of plasma and interstitial fluid. The major contribution to oncotic pressure under physiological conditions is plasma albumin concentration. The hydrostatic pressure of the plasma across the afferent capillary membrane creates a counter force to the oncotic pressure: the combination of the changing hydrostatic and oncotic pressure gradients across the capillary bed is known as the Starling forces. Because the total solute content of cells under physiological conditions is essentially fixed and water is freely permeable across most cell membranes, the volume of the ICF is determined by the body water content. Intracellular fluid tonicity will in turn determine ECF tonicity, but ECF volume is essentially dependent on its sodium content. The sodium content of a normal adult is 55–65 mmol/kg body weight. The concentration of sodium in plasma is ~ 140 mmol/L (~ 152 mmol/kg). Under physiological conditions, the control of the ECF volume is through the control of functioning or effective plasma volume (that part of the plasma volume actively perfusing tissues). There are a variety of afferent mechanisms to monitor effective plasma volume (and thus ECF volume), which include intrathoracic volume receptors such as atrial stretch receptors, hepatic volume receptors, arterial baroreceptors, intrarenal baroreceptors and, possibly, tissue receptors monitoring tissue perfusion. Whatever the actual or relative function of all these sensory systems, their resultant influence is fine control over the renal conservation of sodium and the appetite for oral sodium intake. Sodium intake varies considerably between different human cultural and ethnic groups. Variations in intake between 5 and 500 mmol/24 h have been recorded and physiological mechanisms balance this with the renal excretion of sodium. Sodium delivery to the renal tubules is a function of plasma sodium concentration and the glomerular filtration rate (GFR). Every 24 h, the kidneys of an average healthy adult male will filter in excess of 24 000 mmol of sodium, most of which is reabsorbed in the tubules so that, in health, sodium balance is achieved. In a healthy individual, renal sodium conservation can be extremely efficient, with urine sodium concentration falling to < 1 mmol/L urine. Conversely, when sodium intake is excessive, the capacity to excrete sodium can result in urine sodium concentrations up to 300 mmol/L urine. Under normal physiological conditions, approximately 80% of the sodium in the glomerular filtrate is reabsorbed in the proximal tubules. The protein concentration of the blood within the post-glomerular peritubular capillary beds is believed to exert a strong oncotic pressure on fluid in the proximal tubules, and this in turn helps to regulate the volume of fluid reabsorbed. This process contributes to the autoregulation of filtration and reabsorption known as glomerulo–tubular balance. There has been considerable physiological interest in the control of proximal tubular sodium reabsorption and other intrinsic renal control mechanisms, such as redistribution of filtering activity from superficial nephrons (relatively salt-losing) to juxtamedullary nephrons (relatively salt-retaining). However, to date, the major humoral influences on sodium reabsorption appear to reside in the distal tubules and collecting ducts. Aldosterone is a steroid hormone released from the zona glomerulosa of the adrenal cortex. The major control of aldosterone secretion is through angiotensin II, an octapeptide produced in the circulation as a final product of the action of renin. Renin is a proteolytic enzyme secreted by a group of cells (the juxtaglomerular apparatus) situated between the afferent and efferent glomerular arterioles, and specialist chemoreceptor cells found within the distal convoluted tubular epithelium of the kidneys – the macula densa. Renin release is stimulated by decreased sodium delivery to the distal tubules (sodium depletion, ECF volume contraction), renal artery hypotension (due to systemic hypotension or renal artery stenosis) and by sympathetic nerve activity via β1-adrenergic receptors. The substrate for renin is angiotensinogen, an α2-globulin produced by the liver. Renin releases an amino-terminal decapeptide from angiotensinogen known as angiotensin I, which in turn is acted upon by angiotensin-converting enzyme (ACE), predominantly within the pulmonary capillaries. The action of ACE is to cleave the carboxy-terminal dipeptide of angiotensin I to produce angiotensin II. The renin–angiotensin–aldosterone axis is summarized in Figure 4.1. The major physiological influences on aldosterone are body sodium content and renal perfusion pressure, although hyperkalaemia can stimulate aldosterone release directly. FIGURE 4.1 The renin–angiotensin–aldosterone system. +, stimulatory signal; −, inhibitory signal; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide. Aldosterone acts through the specific nuclear mineralocorticoid receptor, which is protected from cortisol (for which it has equal affinity) through the intracellular production of 11β-hydroxysteroid dehydrogenase (11β-HSD). This enzyme converts cortisol to cortisone, for which the receptor has weak affinity. The response within the principal cells lining the distal tubules and collecting ducts is the apical influx of sodium via epithelial Na+ channel (ENaC) stimulation and its efflux via basolateral Na+,K+-ATPase. The net effect is active sodium reabsorption in exchange for potassium. In addition, angiotensin II has direct vasoconstrictive actions, thus having an immediate influence on effective plasma volume. Glomerular filtration and the action of aldosterone do not constitute the complete control over renal sodium excretion within mammalian systems. The existence of a third factor (or factors) had been proposed for 20 years prior to the identification of a specific natriuretic factor in 1981. This factor was originally identified in rat cardiac atria and was termed atrial natriuretic factor (now known as atrial natriuretic peptide, ANP). Since then, a further two natriuretic peptides have been identified in humans. Circulating ANP is a 28 amino acid (AA) peptide with a 17 AA ring structure formed by a disulphide bridge between cysteines at positions 7 and 23: the gene for the ANP precursor molecule is located on the short arm of chromosome 1. Three exons code for a 151 AA peptide (preproANP) which, following removal of the signal peptide, results in a 126 AA peptide (proANP) – the main storage form. On secretion into the circulation, proANP is cleaved into the N-terminal 1–98 peptide (NT-proANP), and the biologically active 99–126 peptide (ANP). The major stimulus to the secretion of ANP is atrial stretch and the major sites of synthesis are the atria. In 1988, a second natriuretic peptide was identified, in porcine brain, and termed brain natriuretic peptide (BNP). It was later shown to be produced predominantly in the ventricles of the heart. Circulating BNP is a 32 AA peptide, also with a 17 AA ring structure formed by a disulphide bridge between cysteines at positions 10 and 26. The ring structure has a high level of homology with the ring of ANP. Also like that of ANP, the gene for BNP is located on the short arm of chromosome 1. Three exons code for a 134 AA peptide (pre-proBNP) which, following removal of the signal peptide, results in a 108 AA peptide (proBNP). Further cleavage on release into the circulation results in the N-terminal 1–76 peptide (NT-proBNP) and the biologically active 77–108 peptide (BNP). Both ANP and BNP share a common receptor that mediates a natriuretic response in the kidneys by causing an increase in glomerular filtration rate and blocking sodium reabsorption in the inner medullary collecting ducts. They also influence sodium reabsorption through antagonism of the renin–angiotensin–aldosterone axis. Both ANP and BNP reduce sympathetic tone in the peripheral vasculature. The basic physiology of ANP and BNP is summarized in Figure 4.2. FIGURE 4.2 The physiology of ANP and BNP. N-terminal pro-peptides are co-secreted with the active natriuretic peptides. Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) share a common receptor to increase glomerular filtration rate (GFR) and urine sodium excretion (UNaV), and to reduce renin and aldosterone secretion. In 1990, a further ‘natriuretic peptide’ was described in porcine brain and termed C-type natriuretic peptide (CNP). The gene for CNP is coded on chromosome 4. Although CNP has been identified in human plasma, this peptide is mediated through a different receptor and does not have direct natriuretic function, but acts primarily as an antiproliferative regulator in the vascular cell system and as a neuropeptide. There is no doubt that ANP, and to a lesser extent BNP, has important physiological functions in relation to sodium balance. Simple experiments can readily demonstrate changes in ANP and BNP concentrations in relation to dietary sodium intake, and the interaction of ANP and BNP with the renin–angiotensin–aldosterone axis results in a dual ‘fine-tuning’ system of sodium control, using afferent information obtained from the heart and kidneys simultaneously. However, unlike the situation with the renin–angiotensin–aldosterone axis, no primary disorders of natriuretic hormone excess or deficiency have yet been identified with certainty. Because the major stimulus to the release of ANP and BNP is cardiac wall stretch, the major current clinical utility of measuring these peptides, particularly BNP and NT-proBNP, is in the diagnosis and monitoring of cardiac failure. The renal conservation of sodium is remarkably efficient, but when sustained, non-renal losses occur under physiological conditions, such as through sweating due to prolonged vigorous physical activity or as a result of prolonged exposure to high ambient temperature, then a mechanism for increasing sodium intake comes into play – the salt appetite. That such a mechanism exists in humans can be observed in pathological states of impaired sodium conservation such as Addison disease. However, accurately defining the sodium appetite in humans under physiological conditions is a subjective and difficult task. Salt intakes are largely conditioned by traditions of food intake and cultural habits of seasoning food with salt. The impulse to add salt to prepared food appears to be automatic in many people, who do so even before tasting. Animal experiments have implicated an entirely separate, brain-based, renin–angiotensin–aldosterone system as a controlling influence on active salt-seeking behaviour. Water crosses cell membranes by simple diffusion through the lipid bilayer and through specific water channels known as aquaporins. The existence of specific water channels had been suspected for many years, not least to explain the dramatic changes that can occur in water permeability in the epithelial cells of the renal collecting ducts during hydration or dehydration. However, it was not until 1992 that the first aquaporin was characterized in human red cells – aquaporin 1 (AQP 1). To date, 13 different mammalian aquaporins have been identified (AQP 0–12). Specific human disease has been associated with mutations in the genes coding for AQP 0 (congenital cataracts) and AQP 2 (autosomal recessive nephrogenic diabetes insipidus – see p. 41). Although mutations in the gene for AQP 1 have been described, affected individuals remain asymptomatic. In addition, the condition neuromyelitis optica (NMO), a rare demyelinating and inflammatory disease of the central nervous system (CNS), is caused by autoantibodies directed against the main water channel of the CNS – AQP 4. The regulation of the gene expression of aquaporins may have important pathophysiological consequences in disease states associated with water retention or cerebral oedema. Under physiological conditions, the solute content of cells is constant and, therefore, cell volume is dependent on solvent, not solute, content. The majority of cells behave as ‘effective’ osmotic meters – swelling when body water increases and contracting when body water decreases. Normally, the ECF and hence ICF osmolality are maintained at about 285 mmol/kg. There is a minimum obligatory loss of water by the kidneys each day that is dependent upon the maximum achievable urine concentration and the osmotic load for excretion. The maximum renal loss of water occurs when, for a given osmotic load, the minimum urine concentration is achieved. The control of water output by the body is through secretion of antidiuretic hormone (arginine vasopressin, AVP) and its renal action. Arginine vasopressin is a nonapeptide synthesized in magnicellular neurons within two paired nuclei in the hypothalamus – the supraoptic and paraventricular nuclei. The gene for AVP in humans is located on the short arm of chromosome 20. Three exons code for a pre-pro-vasopressin that, following removal of the signal peptide, results in pro-vasopressin, which is subsequently packaged into neurosecretory granules. The granules are then transported by axonal flow to nerve terminals in the posterior pituitary. During transport, pro-vasopressin is further cleaved to AVP, neurophysin II and a glycoprotein. The neurophysin II forms tetramers, with one AVP molecule bound to each neurophysin moiety and the whole possessing a further AVP binding site. The stimulus to release AVP into the circulation results in the simultaneous release of AVP, neurophysin II and the glycoprotein. Closely associated cells within the hypothalamus (the osmoreceptor cells), by virtue of their swelling or shrinking in response to changes in ECF osmolality, control the release of AVP from the posterior pituitary. The effect of changing plasma sodium concentration (and hence osmolality) on plasma AVP concentration is shown in Figure 4.3. Other solutes confined to the ECF, for example exogenously administered mannitol, have a similar effect. By contrast, urea produces no significant stimulation of AVP because it freely permeates cell membranes. The osmoreceptor response is often characterized by its set point (variously defined by the plasma osmolality at which a measurable AVP response commences or by the basal state osmolality) and by its responsiveness (gain or sensitivity as judged by the slope of the response). Thus, in situations of water depletion, ECF osmolality will increase, osmoreceptor cells will contract and plasma AVP secretion will increase. Hydration will reverse these events and suppress AVP. This system constitutes the osmoregulatory control of AVP release. FIGURE 4.3 Osmoregulation of arginine vasopressin (AVP) and thirst. In addition to osmoregulation, certain non-osmotic controls over the secretion of AVP exist, including ECF hypovolaemia, hypotension, nausea and an oropharyngeal response. The AVP response to hypovolaemia and hypotension is relatively insensitive when changes are proportionally small (5–10% reductions), but increases exponentially as further reductions occur. Thus, a reduction in ECF volume or blood pressure of 20% or more will result in a plasma AVP concentration far in excess of that observed during normal osmoregulation. The influence of baroreceptor or volume receptor afferent input appears to modulate the osmotic response but does not abolish it: modulation occurs by decreasing the threshold for AVP release and increasing the gain of the systems. Nausea is the single most powerful stimulus to AVP secretion. It overrides osmoregulatory control, and plasma AVP concentrations may increase 100-fold or more. A short-lived oropharyngeal suppression of AVP can occur following oral ingestion of fluid and prior to any reduction in serum osmolality, thus direct studies of osmoregulation of AVP need to avoid any such stimulus. The apical (luminal) surface of the specialized cells lining the collecting ducts is essentially impermeable to water, except when AVP occupies its specific receptors on the basolateral (contraluminal) surface – the V2 receptor (AVPR2). Stimulation of AVPR2 results in AQP 2 channels, located in vesicles beneath the apical membrane, fusing with that membrane and rapidly delivering water channels to the cell surface. A schematic representation of this model is shown in Figure 4.4. Under these conditions, water is absorbed into the collecting duct cells as a result of medullary hyperosmolality and is thence absorbed into the bloodstream: concentrated urine is formed. During states of overhydration, when AVP secretion is suppressed, the luminal surfaces of the collecting duct cells remain impermeable to water. Luminal fluid rendered hypotonic in the diluting segments of the nephrons is not exposed to the medullary hypertonicity and, hence, no water is absorbed into the bloodstream; dilute urine is formed. The renal response to AVP is thus dependent on an intact receptor–effector mechanism within the collecting duct cells, resulting in an alteration in luminal membrane permeability, and upon the presence of a renal medullary osmotic gradient. Human urine osmolality varies between approximately 50 and 1400 mmol/kg. FIGURE 4.4 Renal action of arginine vasopressin (AVP). Interaction of AVP with its V2 receptor located on the basolateral membrane results in the generation of cAMP, which subsequently causes the fusion of AQP 2 laden vesicles with the otherwise impermeable apical membrane. Under the influence of the medullary osmotic gradient, water is drawn across the cell and is taken up within the blood via AVP-independent AQP 3 water channels. The physiological stimulus to water intake is thirst. However, the act of drinking in human societies in temperate regions is predominantly a social or habitual act not dependent on thirst. The control of water balance under non-pathological conditions is thus, in many individuals and for much of the time, achieved by control of output. The osmoregulation of water output by the kidneys can only modulate and restrict an established deficit, but to reverse such a deficit, a specific water input mechanism – thirst – is required. The osmoregulation of thirst is similar in principle to that of AVP and appears to be primarily governed by alteration in osmoreceptor cell size. The osmoreceptor cells controlling thirst are considered to be closely linked to, but distinct from, those controlling AVP secretion. A common method of study in human subjects is the measurement of subjective thirst during continuous osmotic stimulation with hypertonic saline infusion, and a typical range of normal responses is shown in Figure 4.3. Controversy exists as to the exact relationship between the onset of effective renal water conservation and the onset of effective thirst – that is, a sensation of thirst sufficient to cause active seeking and intake of water. However, physiologically, the onset of AVP secretion (and hence water conservation) differs from the onset of thirst and normally precedes it. The magnitude of this difference in water output and input effector controls will determine whether an individual controls physiological water balance primarily by thirst and water intake or by renal conservation of water. Non-osmotic thirst occurs when extracellular fluid is lost without corresponding cellular dehydration, the osmotic pressure of the extracellular fluid remaining unchanged. In this respect, the overall control of thirst parallels the control of AVP, with both osmotic and hypovolaemic stimuli. There is good evidence from animal experiments of both neural and hormonal mediators controlling non-osmotic thirst. Angiotensin II is the most potent human thirst stimulant and may act directly upon the brain, but even when the effects of angiotensin II are blocked, significant hypovolaemia will still stimulate thirst. Thirst following haemorrhage is a commonly reported clinical observation but, like the AVP responses to extracellular hypovolaemia, often a considerable degree of haemorrhage (15–20% of total blood volume) is necessary before the sensation becomes strong. Thus for day-to-day water balance, the primary physiological control of thirst is osmotic. In health, plasma potassium concentrations range between 3.1 and 4.6 mmol/L, with values in serum approximately 0.3–0.4 mmol/L greater, owing to release of potassium during clot formation (mean serum concentration 4.0 mmol/L). The intracellular concentration of potassium is ~ 160 mmol/kg, and 98% of the total body potassium is present in the intracellular fluid. There are two aspects to the physiological control of potassium, namely, the total body content and its distribution between intra- and extracellular spaces. The potassium content of cells is determined by the balance of activity between uptake of potassium due to membrane-bound Na+,K+-ATPase and the passive loss or leakage of potassium out of the cell. Many factors can influence the distribution of potassium, for example acid–base status, hormones (insulin, catecholamines), osmolality and the cellular content of potassium. The influence of acid–base status is widely recognized as an important contributor to potassium distribution, with an association between hypokalaemia and alkalosis and between hyperkalaemia and acidosis, particularly when the acidosis is induced by mineral rather than organic acids. Insulin promotes active uptake of potassium by cells, probably by direct stimulation of Na+,K+-ATPase, and this activity appears to be independent of the effect of insulin on glucose uptake. The importance of the effects of insulin in controlling plasma potassium under physiological conditions is not understood, but its action has an important therapeutic role in the treatment of hyperkalaemia. Catecholamines have an effect on potassium distribution, with β-adrenergic actions essentially promoting cellular uptake and α-adrenergic actions resulting in increases in plasma potassium concentration, but again the significance of these effects under physiological conditions is not understood. The net effect of catecholamines on cellular uptake of potassium probably explains the transient hypokalaemia frequently observed in acutely ill patients. An acute increase in extracellular tonicity, such as occurs following hyperosmotic infusions of saline or mannitol, results in an increase in plasma potassium concentration. This results from leakage of potassium from cells, a phenomenon that is not related to extracellular acidosis, but may be linked to cellular dehydration, altered cell membrane function or altered cell metabolism. An increase in extracellular tonicity is also observed in patients with hyperglycaemia in the absence of insulin and has important therapeutic relevance in the provision of potassium replacement during the treatment of hyperglycaemia. The effects of tonicity under physiological conditions are probably of no significance. Potassium depletion results in a greater loss of potassium from the ECF than the ICF, and potassium excess results in a greater proportional rise of ECF than of ICF potassium concentration. The controlling influences over these changes are not defined, but the result is a significant alteration in membrane potential: this is increased with potassium depletion and decreased with excess. The effects on neuromuscular function of either condition constitute the most important clinical complications of disorders of potassium metabolism. The traditional understanding of potassium handling by the kidneys is that potassium is freely filtered by the glomeruli, but that up to 95% has been reabsorbed before the tubular fluid reaches the distal convoluted tubules. The predominant control of potassium excretion appears to reside in the control of distal tubular reabsorption and secretion. Plasma potassium itself has a major effect on potassium secretion in the distal tubules, tending to correct any imbalance. Acute changes in sodium delivery to the distal tubules may also influence potassium excretion – restricted sodium delivery impairs potassium excretion, but a tendency to natriuresis is accompanied by a kaliuresis. However, chronic effects on potassium excretion as a result of changes in sodium intake are not seen because of the influence of the renin–angiotensin–aldosterone axis. Potassium directly influences aldosterone secretion from the adrenal cortex. A high plasma potassium concentration stimulates aldosterone secretion and a low concentration suppresses secretion. In addition to its effects on sodium reabsorption by the principal cells, aldosterone stimulates hydrogen ion secretion by the α-intercalated cells of the distal tubules and collecting ducts. Acidosis is associated with reduced potassium secretion and alkalosis with enhanced secretion. The net effect of aldosterone is to stimulate exchange of potassium and hydrogen ions for sodium ions. Therefore, the relative proportions of potassium and hydrogen ions within the cells of the distal tubules, together with the ability to secrete hydrogen ions, will determine the effect of systemic acidosis or alkalosis on potassium excretion. Acting alone, an acidosis will promote potassium retention and an alkalosis will promote a kaliuresis. Urine potassium concentration can vary between about 5 mmol/L and 150 mmol/L. Adaptation of urinary excretion to a variation in input tends to be slow, taking a few days to achieve a new balance. In this respect, urinary control of potassium is less sensitive than the control of sodium. As sodium is predominantly an extracellular cation, the control of sodium balance will control the volume of the ECF. The tonicity of body fluids is under osmoregulatory control, therefore sodium deficit or sodium excess presents clinically with primary changes in ECF volume rather than changes in sodium concentration within the ECF. Hyponatraemic and hypernatraemic states are discussed in the section on water metabolism. Sodium is always lost from the body in association with water. As the sodium concentration of all body fluids is equal to or less than plasma (except on occasions of high sodium intake, when urine sodium concentration may exceed plasma concentration), loss of any body fluid except plasma will generally result in an excess loss of water over sodium. Any loss of sodium, however, will result in a reduction of ECF volume, including a reduction in circulating plasma volume. Clinical presentation will depend on the severity of the decrease. When the changes are mild, patients are often described clinically as being dehydrated, a description that should be confined to pure water deficiency but, unfortunately, in general usage is not. Except in rare instances, truly dehydrated (water depleted) patients are extremely thirsty; patients with all but the most severe salt and water deficiency are not. Reduced intravascular volume, when mild, will result in postural hypotension and a compensatory increase in pulse rate; central venous pressure is reduced and this can be assessed clinically by observation of neck vein filling or directly measured following insertion of a cannula into a central vein. When the volume reduction is more severe, hypotension and eventually shock will result in oliguria; the central venous pressure is further reduced. Reduction in interstitial fluid volume results in reduced skin turgor and transcellular fluid reductions result in dry mouth and reduced intraocular pressure. The causes of sodium deficiency can be classified broadly into extrarenal, primary renal (resulting from renal disease) and secondary renal (resulting from disturbed hormonal control of renal sodium retention or from inappropriate use or abuse of diuretics). In addition, and somewhat difficult to classify, is the sodium deficiency that can occur when isolated jejunal segments are incorporated into urinary diversion operations (jejunal conduit, jejunal continent diversion, see p. 55). These procedures are now rarely performed, as alternative sources of donor intestine are preferred. When jejunum is used in such procedures, there is a risk of postrenal loss of sodium. Extrarenal fluids have sodium concentrations that may approach the concentration in plasma (Table 4.3). The major causes of extrarenal sodium deficiency are summarized in Box 4.1. The commonest clinical presentations result from gastrointestinal disease. The clinical history may considerably underplay the degree of deficit, especially in chronic conditions or conditions resulting in sequestration of fluid. The major causes of primary renal sodium loss are summarized in Box 4.2. The recovery phase of acute kidney injury is often associated with a polyuria, kaliuresis and natriuresis. Usually this stage is short lived, lasting only a few days, but it may occasionally be prolonged. A natriuresis may occur following successful renal transplantation and this may be in part due to transient tubular dysfunction; the recovery phase usually lasts only a few days, but occasional patients show prolonged natriuresis. Relief of urinary tract obstruction, most commonly seen in patients with prostatic enlargement, is often followed by a short period of diuresis and natriuresis. The exact mechanism of this is not fully understood, but is probably related to a urea-induced osmotic diuresis, together with elimination of excess sodium retained during the obstruction phase. The natriuresis in these cases is corrective and is not strictly a primary renal condition: it is unlikely to lead to sodium depletion. Normally, this phase of post-obstructive natriuresis and diuresis lasts between one and seven days, but occasionally a more prolonged natriuresis occurs, leading to sodium deficiency. This rare event is secondary to tubular damage occurring during obstruction. Salt wasting has been described in association with meticillin-induced acute interstitial nephritis. Full resolution of the condition may be delayed for several months, during which time minimum obligatory losses of sodium may exceed 100 mmol/24 h. Chronic kidney disease is generally associated with a reduced capacity to excrete sodium. Considerable adaptation occurs to increase the natriuretic capacity of the remaining functioning nephrons, which paradoxically impairs the overall sodium conserving function. On a Western type diet, sodium intake normally exceeds minimum obligatory urine sodium loss, but if dietary salt is restricted or non-renal losses of sodium are increased, the obligatory loss may induce sodium deficiency. Minimum obligatory losses may be as little as 40 mmol/24 h and even lower if enough time is given for adaptation, but occasionally patients have minimum obligatory losses exceeding 150 mmol/24 h (3 mmol/kg body weight in children). The term salt-losing nephropathy is appropriate for this group of patients. Salt-losing nephropathy is a clinical state, rather than a specific disease. It is normally associated with chronic kidney disease due to tubulointerstitial disease or to glomerulonephritis with significant interstitial abnormalities. A common example of a toxic nephropathy inducing a salt-losing state is analgesic abuse, in which there is typically also reduced concentrating ability and renal tubular acidosis. The disruption of extrinsic controls over renal sodium handling can give rise to a secondary renal sodium loss. Patients present with a variety of symptoms, some specifically related to a contracted ECF volume, such as postural hypotension and an increased sodium appetite. Causes of secondary renal sodium loss, presented in Box 4.3, are essentially hormone or diuretic induced. In Addison disease, the synthesis and secretion of aldosterone are reduced because of adrenal gland destruction, leading to distal renal tubular sodium wasting. Congenital adrenal hyperplasia (CAH) with associated sodium wasting is a result of impaired mineralocorticoid synthesis, most commonly as a consequence of 21-hydroxylase deficiency, but also, less commonly, 3β-hydroxysteroid dehydrogenase deficiency. In either condition, the degree of sodium wasting is variable: it is present in about two-thirds of patients with 21-hydroxylase deficiency (see Chapter 18). Very rare forms also exist, including cholesterol desmolase deficiency (lipoid adrenal hyperplasia) and corticosterone methyl oxidase deficiency – a deficiency in the mixed-function oxidase catalysing the final steps of aldosterone synthesis. Corticosterone methyl oxidase deficiency is not strictly a type of CAH, as the synthesis of cortisol is unaffected and, consequently, the adrenals are not hyperplastic. In all these conditions, the associated hypovolaemia stimulates renin production, and all may be associated with hyperkalaemia. Treatment is primarily by glucocorticoid and mineralocorticoid replacement. It might be predicted that deficient production of renin would also lead to renal sodium loss because of a secondary deficiency of aldosterone. The condition hyporeninaemic hypoaldosteronism is invariably associated with renal insufficiency, but the characteristic feature is hyperkalaemia rather than hypovolaemia. As in the majority of patients with renal insufficiency, inappropriate renal sodium loss will induce hypovolaemia if the patient is placed on a low sodium intake, but it is unclear if this is more severe in hyporeninaemic hypoaldosteronism. Although this condition is a cause of secondary renal sodium loss, its main importance is in the differential diagnosis of hyperkalaemia and it is discussed in more detail later in this chapter (see Syndromes of hypoaldosteronism, p. 60). Pseudohypoaldosteronism (type 1) is a rare congenital disease existing in two forms, caused by an end organ failure of response to aldosterone within the principal cells lining the renal collecting ducts. It is caused by either a loss of function mutation in the mineralocorticoid receptor (MR) gene (autosomal dominant form) or a mutation resulting in loss of function in the epithelial Na+ channel (ENaC – autosomal recessive form). Infants present with dehydration, hyponatraemia, hyperkalaemia, metabolic acidosis, failure to thrive and weight loss. Renal sodium wasting is unresponsive to exogenous mineralocorticoids. Pseudohypoaldosteronism is also discussed in more detail later in this chapter (see Syndromes of hypoaldosteronism, p. 60). Diuretic abuse, especially when surreptitious, may present a difficult diagnostic problem. The major potent diuretics, such as furosemide or bumetanide, may induce significant renal sodium loss and the differential diagnosis includes Bartter syndrome. Chronic abuse with thiazide diuretics may produce a clinical picture similar to Gitelman syndrome. In all cases, however, the major and persistent finding is significant hypokalaemia (see p. 55). No single laboratory finding is diagnostic of sodium deficiency, which, therefore, remains primarily a clinical diagnosis. There may be uraemia, particularly of a prerenal pattern (with urea concentration disproportionately elevated in relation to that of creatinine). Serum sodium concentration may be normal, decreased (usually in association with uraemia) or even increased when the sodium deficit is proportionately less than the water deficit. Serum sodium and urea concentrations cannot indicate accurately the degree of sodium depletion or reduction in ECF volume. Measurement of the haematocrit may help to establish the degree of reduction in ECF volume, especially if the individual’s normal haematocrit is known (see Appendix 4.1a). Similarly, serum protein measurements may suggest ECF volume contraction. In a steady state, hyponatraemia in association with clinical evidence of sodium deficit can also be used to estimate the extent of sodium deficit (see Appendix 4.1b). Either method provides only a rough guide to subsequent replacement. Urine electrolytes will help to establish whether the loss has been renal or extrarenal. If there has been gastrointestinal loss of sodium or another extrarenal loss, renal sodium retention should be maximized. Random urine sodium concentrations will show conservation of sodium with a concentration < 10 mmol/L. The mature, fully functioning renal system can reduce urine sodium concentration to < 1 mmol/L, but this capacity is less in the very young and old. Twenty-four hour urine sodium output will show significant sodium retention, with 24 h urine sodium excretion as low as 1 mmol and invariably < 15 mmol, even in the elderly. Serum osmolality measurements are of no value in the differential diagnosis of sodium depletion, adding nothing to the information obtained from serum electrolytes, urea and creatinine measurements. Urine osmolality will generally show an overall significant increase towards maximum, reflecting the oliguric state, especially when sodium depletion is sufficient to cause a non-osmotic increase in arginine vasopressin (AVP). Only when a renal loss of sodium is present without evidence of renal impairment should secondary hormonal or diuretic-related sodium depletion be considered. For the majority of hypoaldosterone-related disorders, there will be associated hyperkalaemia (for specific investigation of hypoaldosteronism, see Chapter 18). Diuretic abuse can provide an intriguing puzzle as, along with Bartter syndrome and Gitelman syndrome, it provides the major differential diagnosis of renal sodium loss in association with hypokalaemia. The essential steps in treating sodium deficiency and ECF volume depletion are to attempt to treat the causes of sodium and water loss and to adequately replace the fluid already lost. The amount of fluid replacement must be balanced against measured or estimated losses, both intra- and extracorporeal. The measurement of body weight is a useful adjunct to monitoring; an increase in weight may indicate accumulation of interstitial or transcellular fluid. Accurate fluid balance charts (see Table 4.4) should indicate a positive or negative balance, but even the most assiduously prepared charts (many are not) provide only an approximate estimate of balance, especially when confounding problems such as pyrexia (increasing insensible losses) or hypermetabolic states (increasing metabolic water) intervene. Vital signs: pulse, blood pressure and central venous pressure (if measured), all provide additional useful information to improve the practice of the art. TABLE 4.4 Ideal components of a fluid balance chart The type of replacement will be dependent on cause and severity. Mild forms of sodium deficit, such as caused by overtreatment with diuretics or chronic salt-losing nephropathy, may be adequately treated with oral sodium and water supplementation. For more severe forms of sodium deficit, intravenous infusion is required; the types of fluids available are shown in Table 4.5. All of them are iso- or hypoosmolal to normal plasma. Hyperosmolal fluids, such as 1.8% or 5% sodium chloride or 2.74% or 8.4% sodium bicarbonate, are not appropriate treatments for sodium depletion with clinical evidence of reduced ECF volume, even in the presence of hyponatraemia, as infusions of such fluids will increase ECF volume in part by shifting fluid from the ICF volume. Hypoosmolal fluids may be used when sodium deficit is associated with a greater degree of water deficit, that is, sodium deficit in association with hypernatraemia. Sodium excess is almost invariably associated with water excess (and is usually an iso-osmolal sodium excess). The clinical presentation will depend on the severity of the expansion of ECF volume and its relative distribution within the ECF volume compartments. In practice, the clinical presentation is with oedema (peripheral or pulmonary) or effusions (pleural, ascites) or with hypertension. Peripheral oedema, due to sodium retention, is characteristically ‘pitting’ when digital pressure is applied to the affected part (lymphoedema, due to lymphatic obstruction, is characteristically non-pitting, while in myxoedema, the swelling is brawny in nature). Measurement or clinical observation of the central venous pressure may suggest an associated increase in intravascular volume, as may the demonstration of hepatic congestion or the presence of pulmonary crepitations. Hypertension is a common clinical finding, but hypertension specifically due to chronic iso-osmolal sodium excess is rare. Causes of this form of hypertension are discussed later, but all are associated with an increased mineralocorticoid effect in the initial stages, promoting renal sodium conservation and renal loss of potassium. The association of muscle weakness (caused by hypokalaemia) with hypertension is an important clinical pointer to such a cause of sodium retention. The causes of sodium excess in association with oedema formation are shown in Box 4.4. Congestive cardiac failure (CCF) is a common clinical example of sodium excess, presenting with dependent oedema, congested liver, increased jugular venous pressure and pulmonary crepitations due to oedema. The ‘backward failure’ hypothesis suggests that increased transcapillary pressure resulting from increased central venous pressure causes oedema and hence reduces intravascular volume. Reduced intravascular volume will in turn stimulate sodium retention. Although readily understandable, this theory is inadequate because patients generally have increased intravascular volumes. The ‘forward failure’ hypothesis suggests inadequate perfusion of the kidneys because of reduced cardiac output, which in turn promotes sodium retention. However, high cardiac output failure demonstrates that sodium retention can be independent of cardiac output. The reduced ‘effective circulating volume hypothesis’ lacks precise definition, but suggests that only a proportion of the volume is ‘effective’, that is, circulating volume appears to be reduced in that the renin–angiotensin–aldosterone mechanism is activated, as is the release of AVP. The onset of CCF causes ANP and, particularly, BNP, release, leading to markedly elevated plasma concentrations. However, the natriuretic effect of the hormones is severely blunted and corrective natriuresis does not occur without therapeutic intervention, with diuretics or with exogenous natriuretic peptide administration in pharmacological doses. The nephrotic syndrome is characterized by severe glomerular proteinuria, hypoalbuminaemia and oedema. The classic hypothesis explained the oedema and secondary sodium retention, on the basis of reduced oncotic pressure owing to hypoalbuminaemia. However, the rare condition congenital analbuminaemia is not associated with significant oedema, and nephrotic patients infused with albumin do not consistently respond with a natriuresis and reduction of oedema. Sudden remission of minimal-change glomerulonephritis may result in clinical resolution prior to an increase in plasma albumin, and the presumed reduced circulating volume in nephrotic syndrome is less commonly found when sought than normal or increased circulating volume. These observations imply an intrarenal mechanism of sodium retention in the nephrotic syndrome. Sodium retention does not appear to be primarily driven by the renin–angiotensin–aldosterone system; moreover, ANP concentrations are moderately increased, not decreased, and the natriuretic effect of the hormone is maintained. Chronic liver disease frequently results in a state of sodium retention leading to oedema and ascites. Again, the physiology of sodium retention is multifactorial, resulting from decreases in glomerular filtration, enhanced proximal tubular reabsorption and increased renin–angiotensin–aldosterone production, presumably from decreased ‘effective’ intravascular volume. As in CCF, the plasma ANP concentration is significantly increased, but again the natriuretic response is blunted. The concentration of AVP may also be significantly increased. Pregnancy demands a retention of between 700 and 1000 mmol of sodium in order to expand the maternal plasma volume and interstitial space and to provide the fetus with sodium. Sodium retention occurs progressively during pregnancy, even though the glomerular filtration rate is greatly increased, so that the reabsorption of filtered sodium must be substantially increased above the non-pregnant level. The renin–angiotensin–aldosterone system is stimulated during pregnancy, a finding that supports the theory of renal sodium retention as a response to reduced ‘effective’ vascular volume (due to redistribution of ECF volume to the growing fetus and its maternal circulatory support). However, many studies have shown increased plasma ANP concentrations during pregnancy, a finding somewhat at odds with the presumed reduced ‘effective’ vascular volume. Thus, the kidneys may retain sodium in response to some other stimulus (such as the effect of oestrogens), and the increase in ANP may represent the response to the resultant increased atrial pressure from expanded vascular volume. However, these two apparently conflicting theories are not mutually exclusive and it seems likely that the resultant increase both in the activity of the renin–angiotensin–aldosterone system and the secretion of ANP forms the basis for an enhanced control over sodium balance. Enormous differences in daily sodium intake (from < 10 mmol to > 300 mmol) can be adequately accommodated during pregnancy. Dependent leg oedema is extremely common during pregnancy, especially during the third trimester, and much of this is attributable to the mechanical pressure of the uterus upon the venous return from the lower limbs. However, there is a general tendency to an increased interstitial fluid content because of reduced oncotic pressure (as a result of reduced plasma protein concentration) and because of the effects of oestrogens, which enhance the hydration of the mucopolysaccharide ground substance of connective tissue. This general tendency to increased interstitial fluid content may also produce a more generalized oedema. Excessive sodium retention occurs in pre-eclampsia. Total body sodium is increased, but plasma volume is usually decreased, with shifts of fluid to the interstitium. The reduced plasma volume is the result of intense vasoconstriction, which also results in hypertension. In pre-eclampsia, renin activity and aldosterone concentrations in plasma in the third trimester are less than those observed during normal pregnancy. Furthermore, ANP concentrations are significantly increased above those found in normal pregnancy, or indeed those found in pregnancy associated with essential hypertension. Atrial natriuretic peptide appears to be released into the maternal circulation in response to vasoconstriction and an increased volume load to the cardiac atria. Thus in pre-eclampsia, as compared with normal pregnancy, renin, aldosterone and ANP all act as if to promote natriuresis, but, for reasons which are not understood, natriuresis is impaired. Despite intensive study over 50 years or so, the question of whether the normal menstrual cycle is associated with sodium retention remains unresolved. There is widespread belief that the majority of women retain sodium premenstrually, giving rise to premenstrual oedema in some. Plasma aldosterone and renin concentrations are elevated during the luteal phase of the ovulatory cycle, but this elevation is coupled with an increase in progesterone, which is thought to antagonize the renal actions of aldosterone. Studies have shown that plasma ANP concentrations remain the same during the follicular and luteal phases. It has also been demonstrated that, in healthy females, the ANP response to volume expansion is no different in the follicular and luteal phases of the menstrual cycle, and the proportional suppression of renin and aldosterone is also identical between the two phases of the cycle. When these findings are coupled with the observations that body weight, creatinine clearance and basal sodium excretion do not alter during the phases of the cycle, it is apparent that there is little evidence of renal sodium retention. This remains an area for further research, particularly as inappropriate treatment of perceived symptoms may lead to further complications of sodium balance (see below). Idiopathic oedema, sometimes known as cyclical oedema, is a condition that occurs in females postpuberty. Oedema of the face, hands and legs can develop rapidly, and weight gains up to 4 kg in 24 h have been described. The aetiology of this condition is unknown, but one suggestion is that diuretics may be used to produce rapid weight loss and that cessation of this self-medication leads to rebound sodium retention. The diuretic is subsequently recommenced for cosmetic reasons to reduce oedema formation and weight gain. Thus, a vicious cycle of sodium depletion followed by sodium retention and oedema is set in place. However, in some patients prior diuretic use cannot be demonstrated. Many patients with idiopathic oedema show exaggerated sodium retention on assuming an upright posture with a marked reduction in renal blood flow and GFR. Oestrogens do not appear to play an important role, as the condition has been demonstrated in a patient who had previously undergone bilateral oophorectomy. The role of renin–angiotensin–aldosterone is not fully understood, with a proportion of patients having increased plasma renin and aldosterone concentrations, particularly when in an upright posture, but with suppression of concentrations by a high salt diet. Limited studies of ANP have been performed, but no abnormalities have been demonstrated: normal basal values are found and are stimulated by volume expansion. The causes of sodium excess without oedema are shown in Box 4.4. Acute sodium loading is a rare event, invariably caused by inappropriate sodium administration to highly dependent individuals, for example those in intensive therapy units, especially the very young, the old and the highly incapacitated. Acute hypernatraemia is a powerful stimulus for thirst and thus, to maintain the condition, the intake of fluid must be prevented by an inability, for whatever reason, to express or act upon the desire to drink. Examples of acute sodium loading include administration of high sodium concentration oral feeds to infants (either accidentally or as a form of abuse), the use of oral hypertonic sodium chloride solutions as emetics, excessive administration of intravenous hypertonic sodium bicarbonate and the voluntary consumption of excessive quantities of table salt. In the past, the accidental introduction of hypertonic sodium into the circulation during therapeutic abortion has been a cause of acute sodium excess. Acute sodium loading associated with hypernatraemia is a life-threatening condition and should be treated promptly with free water administration. Mineralocorticoid excess has a variety of causes (see Box 4.4). Although this is a condition of excess sodium retention, the usual clinical presentation is hypertension with hypokalaemia. The severity of hypokalaemia is in turn dependent on sodium intake, being reduced in severity if sodium intake is reduced. All of these conditions are discussed elsewhere in this book, either as part of the pathology of the adrenal glands (see Chapter 18) or in relation to hypokalaemia (see p. 55). The fascination of these conditions with respect to sodium is why sodium retention does not usually progress to increase the volume of all the ECF spaces sufficiently to cause oedema. As the ECF volume expands, the filtered load of sodium increases and the fraction reabsorbed by the renal tubules decreases. This mechanism constitutes the ‘escape’ for sodium (but not potassium) from mineralocorticoid effects. The underlying mechanism of escape has been the subject of investigation since it was first described over 50 years ago. The mechanism appears multifactorial with a haemodynamic response to increased intravascular volume resulting in reduced proximal reabsorption of sodium. There is good evidence that natriuretic peptides may account for a part of the escape mechanism – ANP concentrations are elevated in primary hyperaldosteronism and return to normal when the effects of hyperaldosteronism are reversed or antagonized. Evidence also exists of a decrease in the thiazide-sensitive Na+-Cl− co-transporter (NCCT) within the distal convoluted tubules. In addition intrarenal substances such as prostaglandins, kinins and nitric oxide may also promote natriuresis. The investigation of sodium excess is confined almost exclusively to the investigation of oedema and of hypertension; hypernatraemia is not a feature of sodium excess except in rare instances of acute sodium loading, when the cause is usually clear from the clinical history. In practice, the purpose of the investigation of oedema is primarily to differentiate reduced oncotic pressure from other causes, usually by the measurement of serum protein concentration and the confirmation of any route of protein loss, for example the measurement of urine protein excretion. The diagnosis of oedema secondary to CCF and liver disease is primarily clinical; detailed sodium balance studies are not normally justified or, indeed, practicable. Idiopathic oedema is also a condition that is initially diagnosed from the clinical history, although periods of continuous sodium loading with simultaneous sodium balance studies may reveal a propensity for sodium retention. The laboratory investigation of sodium excess as a contributor to hypertension involves the demonstration of increased mineralocorticoid action, the simplest pointer being the association of hypokalaemia and hypertension. Investigation of primary hyperaldosteronism, conditions of increased glucocorticoid secretion and conditions of sodium retention due to congenital adrenal hyperplasia are covered in Chapter 18. Other causes of hypokalaemia and hypertension are discussed in the section on potassium metabolism (see p. 55). For those conditions associated with oedema, the management will be aimed primarily at ameliorating the primary cause. This primary treatment, when applicable, needs to be coupled with attempts to control sodium and water retention by the restriction of dietary sodium and, when appropriate, the use of diuretics. The laboratory’s role in management is to monitor the potential complications of diuretic treatment, notably hypokalaemia (especially with the use of loop diuretics), hyperkalaemia (with the use of potassium-sparing diuretics such as spironolactone) and hyponatraemia (when over-diuresis results in non-osmotic stimulation of AVP with secondary water retention). In addition, the laboratory’s function is to monitor the effects of natriuresis on renal function by monitoring serum urea or creatinine concentrations. For conditions of sodium excess without oedema, the initial management is to increase renal sodium excretion, either by attention to iatrogenic causes (such as exogenous mineralocorticoid treatment) or by treatment with diuretics such as spironolactone. This treatment is combined, when appropriate, with antihypertensive therapy. The laboratory’s role in management is to monitor for possible complications, particularly with respect to potassium homoeostasis. For those patients with surgically correctable conditions, such as primary hyperaldosteronism due to an adrenal adenoma, the definitive treatment is surgery. As water is distributed throughout all body spaces, the effects of pathological excess or deficiency will be reflected in the relative functional sensitivities of each space. For example, a 10% excess or reduction in total body water (TBW) is unlikely to result in clinical features of alterations in ECF volume. However, ICF volume changes may considerably impair cellular function; in particular, rapid changes may significantly impair brain cell function. Disorders of water metabolism include three categories of conditions. First are conditions in which polyuria is the major feature. Polyuria is defined as a urine output in adults in excess of 50 mL/kg body weight/24 h. In polyuria, one aspect of water homoeostasis is defective but may be compensated by another: either a reduced ability to concentrate the urine is compensated by a secondary increase in thirst, or an excessive water intake is compensated with a secondary increase in urine output. In either case, patients are usually normonatraemic unless the compensatory mechanism is compromised. Second are those of water deficiency in which homoeostasis is defective and normal body water content cannot be maintained: patients present with hypernatraemia (serum sodium above 145 mmol/L). Third are conditions of primary water excess in which homoeostasis is defective: patients present with hyponatraemia (usually defined as a serum sodium < 130 mmol/L) or clinical features of water intoxication. Polyuria is an early feature of diabetes mellitus as a result of the osmotic effects of an increased filtered load of glucose. Polyuria can also be a feature of renal failure due to the loss of medullary hypertonicity and the reduction in the production of AQP 2. Thus polyuria due to diabetes mellitus or renal failure should be differentiated at the outset. A primary inability to concentrate urine with secondary polydipsia is known as diabetes insipidus (DI), and may be due to either impaired release of AVP from the posterior pituitary (cranial diabetes insipidus, CDI) or to impaired renal response to AVP (nephrogenic diabetes insipidus, NDI). The major causes of CDI and NDI are shown in Box 4.5. There are four causes of inherited CDI, all extremely rare. Autosomal dominant CDI is caused by mutations involving the pre-pro-vasopressin gene excluding the region coding for AVP itself. The disorder is due to a progressive degeneration of magnicellular neurons, probably due to the accumulation of abnormal AVP-neurophysin II complexes. Presentation may be in the neonatal period or delayed until later in childhood. An even rarer autosomal recessive form exists in which the coding for AVP is mutated; presentation is in the early neonatal period. The two other causes of inherited CDI are Wolfram syndrome and congenital septo-optic dysplasia, with about one-third of patients with each condition exhibiting overt symptoms of CDI. Wolfram syndrome may present with a combination of diabetes insipidus, diabetes mellitus, optic atrophy and deafness – hence the alternative name of DIDMOAD syndrome. Patients with either CDI or NDI may retain a partial ability to concentrate the urine, which can lead to diagnostic confusion. The osmoregulatory set point for AVP release and thirst is maintained in both conditions. Thus, in developing CDI, as the number of active cells releasing AVP from the posterior pituitary diminishes, the slope of the osmoregulatory response will decrease (see Fig. 4.5). With a plasma osmolality for thirst normally set slightly above that for AVP release, the urine concentration that can be achieved at the onset of thirst will diminish. When CDI is severe, thirst will be stimulated even though the urine is close to maximum dilution. Intermediate stages can, therefore, provide confusion, as an increase in plasma osmolality due to water deprivation despite thirst can be associated with a normally concentrated urine. Diagnostic confusion may be further compounded by two additional features not shown in Figure 4.5:
Sodium, water and potassium
PHYSIOLOGY
Introduction
Other predominant intracellular counter anions are sulphates and proteinates
Electrolyte
ECF (mmol/kg)
ICF (mmol/kg)
Sodium
152
10
Potassium
4.3
160
Calcium
2.7
1.0
Magnesium
1.1
13
Chloride
109
10
Bicarbonate
29
10
Phosphate
1.5
50
Extracellular fluid and sodium
Renal control of sodium output
Intrinsic renal control of tubular reabsorption of sodium
Renin–angiotensin–aldosterone axis
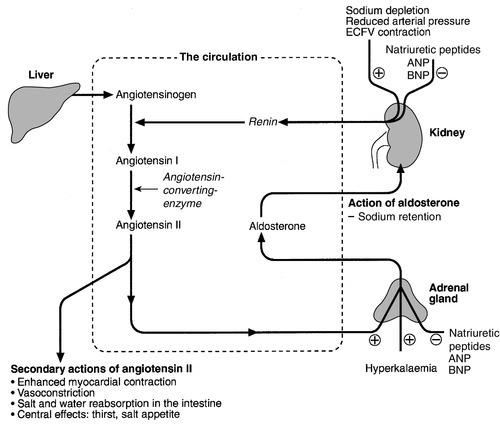
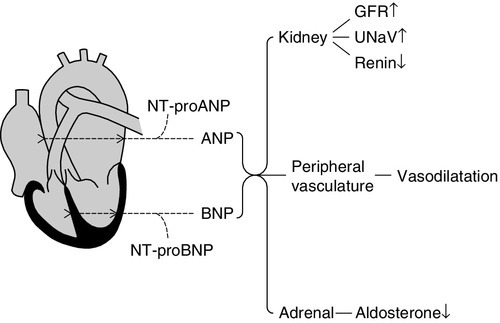
Natriuretic peptides
Sodium appetite
Intracellular fluid and water
Control of renal water output
Osmoregulation
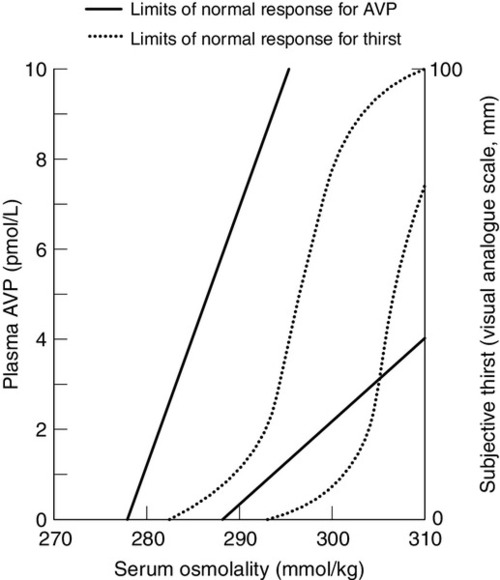
Non-osmotic control of arginine vasopressin
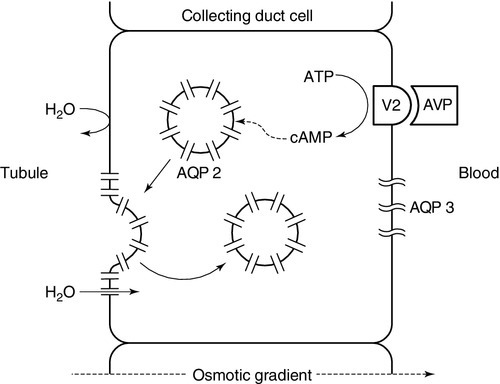
Renal responsiveness to arginine vasopressin
Control of water intake
Osmoregulation
Non-osmotic control of thirst
Extracellular fluid, intracellular fluid and potassium
Extracellular and intracellular fluid distribution of potassium
Renal control of potassium output
Intrinsic tubular control
Aldosterone
DISORDERS OF SODIUM METABOLISM
Sodium deficiency
Clinical presentation
Causes of sodium deficiency
Extrarenal sodium loss
Primary renal sodium loss
Secondary renal sodium loss
Laboratory investigation of sodium deficiency
Management of sodium deficiency
Input
Output
Extravasation
Oral fluid
Urine output
Body weight
Intravenous fluid
Gastrointestinal losses
Water generated in metabolism (~ 500 mL/24 h)
Surgical drains etc.
Insensible losses (~ 900 mL/24 h)
Sodium excess
Clinical presentation
Causes of sodium excess
Sodium excess with oedema
Pregnancy
Menstrual cycle
Idiopathic oedema
Sodium excess without oedema
Laboratory investigation of sodium excess
Management of sodium excess
DISORDERS OF WATER METABOLISM
Polyuria
Primary polyuria with secondary polydipsia
Basicmedical Key
Fastest Basicmedical Insight Engine