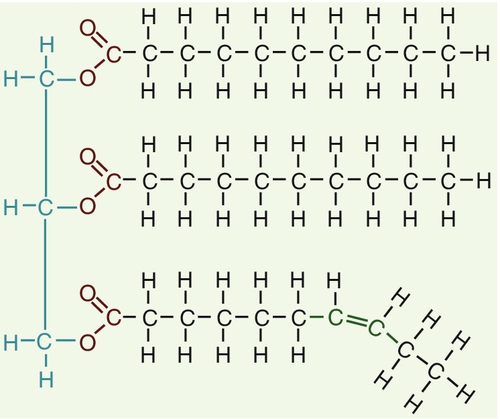
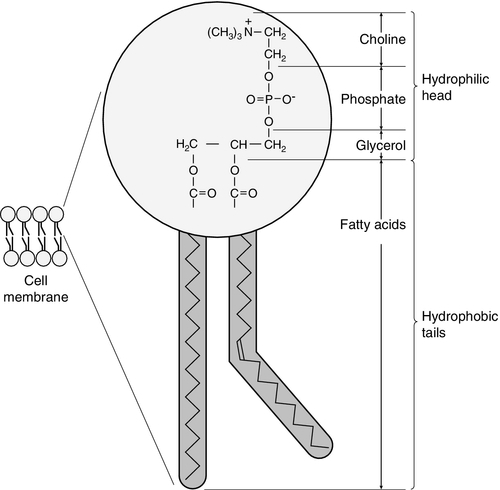
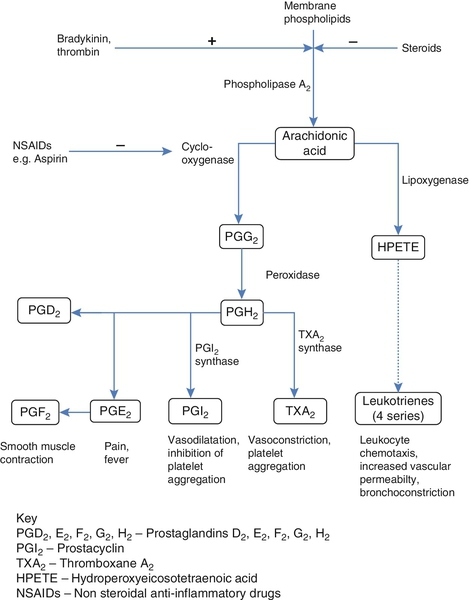
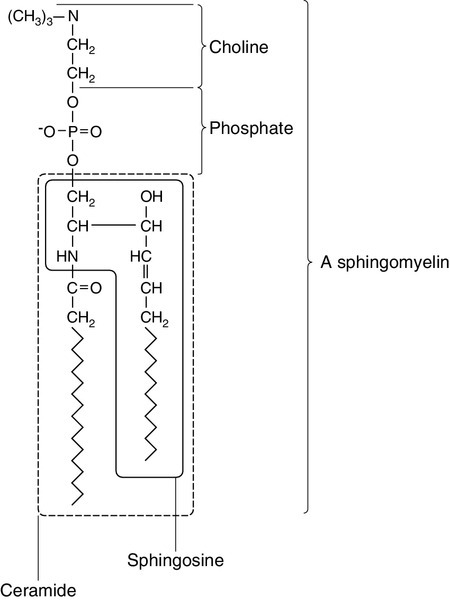
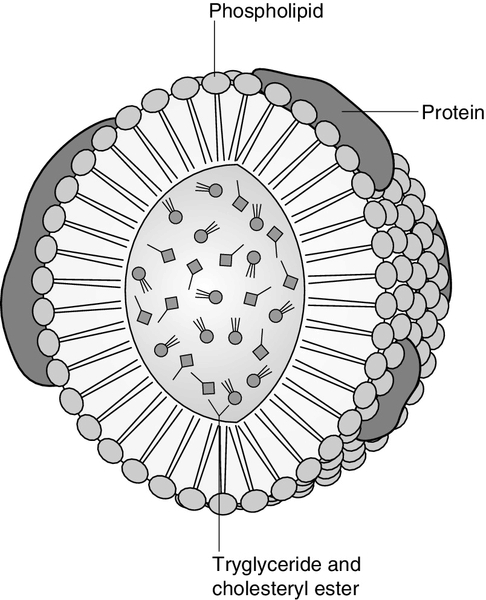
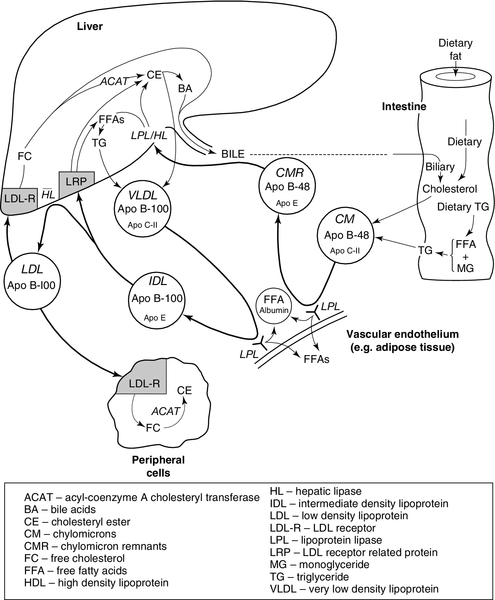
CHAPTER 37 CHAPTER OUTLINE Intermediate density lipoproteins Assembly of apolipoprotein B-containing lipoproteins High density lipoprotein metabolism ENZYMES INVOLVED IN LIPOPROTEIN METABOLISM Lecithin cholesterol acyltransferase Acyl-CoA:cholesterol acyltransferase TRANSFER PROTEINS INVOLVED IN LIPOPROTEIN METABOLISM Cholesteryl ester transfer protein (CETP) Phospholipid transfer protein (PTP) RECEPTORS INVOLVED IN LIPOPROTEIN METABOLISM Scavenger receptor class B type 1 Peroxisome proliferator-activated receptor family OTHER PROTEINS INVOLVED IN LIPOPROTEIN SYNTHESIS, TRANSPORT AND METABOLISM Microsomal triglyceride transfer protein ATP binding cassette transporter family Proprotein convertase subtilisin kexin 9 Sterol regulatory element binding proteins Glycosylphosphatidylinositol-anchored HDL-binding protein 1 CLASSIFICATION OF LIPOPROTEIN DISORDERS THE PRIMARY DYSLIPOPROTEINAEMIAS Familial combined hyperlipidaemia Familial hypertriglyceridaemia Familial hypercholesterolaemia Polygenic hypercholesterolaemia INVESTIGATION OF LIPID DISORDERS High density lipoprotein cholesterol Low density lipoprotein cholesterol Post-heparin lipolytic activity Lipoprotein separation techniques ‘Lipid’ is the term used to describe a number of substances of diverse chemical structure that bear little functional relationship to each other but which have in common the property of being soluble in organic solvents and virtually insoluble in water. Lipoproteins are macromolecular protein complexes that allow hydrophobic lipids to be transported within the hydrophilic environment of the circulation. Lipids are essential for health, but excessive concentrations of cholesterol and triglycerides in the circulation, whether due to lifestyle factors or to inherited disorders of lipoprotein metabolism, are major factors in the development of atherosclerosis and cardiovascular disease. These conditions are the focus of this chapter, although other lipids and their functions are discussed briefly. Lipids can be broadly divided into sterols, including cholesterol; fatty acids or substances containing fatty acids such as triglycerides and phospholipids; eicosanoids; the fat-soluble vitamins (A, D, E and K), and sphingolipids. The major classes of lipids and their principal functions are summarized in Table 37.1. Cholesterol (Fig. 37.1) is the major sterol in humans, being present in all body cells and most body fluids. The majority of the cholesterol in the body is in the free, unesterified form; it is this form that is the structural component of cell membranes. Cholesteryl esters in normal cells represent a store for future use and appear microscopically as intracellular droplets. Cholesterol is present in the diet but most cholesterol in the body is made by de novo synthesis from acetate. The rate-limiting step in the synthetic pathway is the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, catalysed by the enzyme HMG-CoA reductase. The liver is responsible for most cholesterol synthesis. Cholesterol is a precursor for the synthesis of gonadal and adrenal steroid hormones, vitamin D and bile acids. FIGURE 37.1 Structure of cholesterol. Although only a small amount of the body’s cholesterol pool comes from dietary cholesterol, this has an important role in regulating the rate of cholesterol synthesis. The liver is the key organ in maintaining cholesterol balance; any excess cholesterol is excreted by the liver into the bile, either directly, or after conversion into bile acids. Cholesterol is a major component of cell membranes. Cholesterol and sphingomyelin form plasma membrane ‘lipid rafts’ or caveolae. Caveolae are cell surface invaginations found in differentiated cells and characterized by the presence of a protein, caveolin-1; they are sites where signalling molecules are concentrated. In order for these signalling molecules to function, the cholesterol concentration of the plasma membrane must remain constant. This is achieved by a regulatory system that senses the cholesterol content of the membrane and modulates the transcription of genes encoding proteins involved in cholesterol synthesis (e.g. HMG-CoA synthase and HMG-CoA reductase) and cholesterol uptake (e.g. the LDL receptor). The system that does this is a family of membrane-bound transcription factors called sterol regulatory element-binding proteins (see p. 721). Phytosterols are sterols derived by plants; they differ slightly from cholesterol. There are two general types: δ5-phytosterols (e.g. β-sitosterol) and 5α-reduced phytosterols, otherwise referred to as ‘stanols’. In natural foods, the sterols predominate. Both sterols and stanols are incorporated into commercially available foods promoted as cholesterol-lowering agents: they act by competing with cholesterol for absorption from the gut, and may lower plasma cholesterol concentration by 10–15%. Fatty acids (Fig. 37.2) have the general chemical formula RCOOH. Those relevant to human nutrition are the long chain (C12–C20) fatty acids containing even numbers of carbon atoms. They are further defined as saturated, for example stearic (C18:0); monounsaturated, for example oleic (C18:1), and polyunsaturated, for example linoleic (C18:2) and linolenic (C18:3), the second figures indicating the number of double bonds. In general, dietary saturated fatty acids originate from animals and the unsaturated fatty acids from plants. There are, however, exceptions, for example palmitic acid (C16:0) is saturated but derived from palm oil, and the ω-3 series, which are unsaturated but found in fish. The position of the carbon of the first double bond in the polyunsaturated fatty acids differentiates the ω-6 series (where the fist double bond starts beyond the 6th carbon atom from the methyl end of the molecule), from the ω-3 series, where it starts beyond the 3rd carbon atom. FIGURE 37.2 Structure of fatty acids. The long chain fatty acids are oxidized for energy production by a process known as β-oxidation; this results in the sequential shortening of the chain by two carbon atoms and the production of acetyl-CoA. Small amounts of long chain fatty acids are elongated to very long chain fatty acids (VLCFAs), which have a structural function in certain specialized cells. Triglycerides (see Fig. 37.3) comprise three fatty acids esterified with a glycerol backbone. ‘Triacylglycerols’ is the correct chemical name but they are more commonly known as ‘triglycerides’ and this term will be used throughout this chapter. Triglycerides are the major dietary fat. They are hydrolysed in the gut by lipases to fatty acids and monoglycerides. The monoglycerides undergo re-esterification in enterocytes and subsequent incorporation into chylomicrons. The major sites of endogenous triglyceride synthesis are the liver and adipose tissue. In normal circumstances, hepatic triglyceride is secreted in very low density lipoproteins (VLDL). In certain pathological states, triglyceride accumulates in hepatocytes, leading to hepatic steatosis. Adipose tissue triglyceride represents the major energy store of the body. Fatty acids are mobilized from adipose tissue triglycerides by the action of hormone-sensitive lipase (HSL), which is activated by glucagon and adrenaline (epinephrine) and inhibited by insulin. FIGURE 37.3 Structure of triglycerides. Phospholipids, like triglycerides, have a glycerol backbone; in phospholipids, this is esterified with two fatty acids and the third hydroxyl group is linked via a phosphodiester bond to an amino alcohol such as choline, serine or ethanolamine (Fig. 37.4). Phospholipids are therefore amphipathic molecules, with both hydrophilic (phosphate group) and hydrophobic (fatty acid) domains. This property is responsible for the capacity of phospholipids to solubilize other lipids and accounts for their location on the surfaces of lipoprotein molecules and in the cell membrane lipid bilayer. FIGURE 37.4 Structure of phospholipids. This group of compounds takes its name from the systematic name of C20 (eicosa-) fatty acids from which they are derived. It includes prostaglandins, thromboxanes and leukotrienes. These were originally named because they were found in prostate, platelets (thrombocytes) and white cells (leukocytes), respectively. They have major effects on the immune response, reproductive function (including the induction of labour), cholesterol metabolism, smooth muscle function (causing vasoconstriction or dilatation), platelet aggregation and thrombosis. Prostaglandins contain a substituted cyclopentane ring. Thromboxanes are oxygenated eicosanoids closely related to the prostaglandins. The rate-limiting step in the synthesis of both (see Fig. 37.5) is the phospholipase A2-mediated release of fatty acids from phospholipids. The principal precursor of prostaglandins is arachidonic acid (C20:4, eicosatetraenoic acid). The first two steps in their synthesis are catalysed by prostaglandin endoperoxide synthase, which has both cyclooxygenase and peroxidase activity. The major products of this enzyme’s activity are prostaglandin G2 (PGG2) and prostaglandin H2 (PGH2). These are both prostaglandins of the 2 series, having 2 carbon–carbon double bonds. Prostaglandin H2 is converted to thromboxane A2 (TXA2) in platelets and prostacyclin (PGI2) in the arterial wall. Arachidonic acid also serves as a substrate for various lipoxygenases, which generate the leukotrienes, inflammatory mediators and potent stimulators of muscle contraction. Eicosatrienoic acid (20:3) and eicosapentenoic acid (20:5), are alternative precursors for prostaglandin synthesis. The latter generates prostaglandins of the 3 series and leukotrienes of the 5 series. FIGURE 37.5 An outline of eicosanoid synthesis. Eicosanoid imbalance is of interest in atherosclerosis because several recognized risk factors for atherosclerosis, such as smoking, hypertension and diabetes mellitus, are associated with changes in eicosanoid production. Aspirin inhibits cyclooxygenase, thereby reducing prostaglandin, especially platelet TXA2, synthesis. This is the rationale for the use of aspirin in the prophylaxis of atherosclerotic vascular disease. Steroids directly inhibit the release of arachidonic acid from membrane phospholipids. The backbone of sphingolipids is sphingosine or a similar long chain base. The sphingolipids vary in chain length from C14 to C20. Sphingosine itself has 18 carbon atoms and is formed from the condensation of palmitoyl CoA (C16:0) with serine. Another fatty acid chain is joined to sphingosine through an amide link to form a ceramide. Ceramides have a free hydroxyl group that allows reaction with another component. If this contains a phosphate group, then the resultant products are a type of phospholipid called sphingophospholids, also termed sphingomyelins (Fig. 37.6), which are essential components of nerve cells. If a carbohydrate group is attached to ceramide then glycosphingolipids, or simply glycolipids, are formed. These include cerebrosides, sulfatides, globosides and gangliosides. Glycosides of ceramide are referred to as cerebrosides: they are present in relatively high concentrations in brain. Many glycosphingolipids contain oligosaccharides; those that contain more than one molecule of sialic acid are referred to as gangliosides. FIGURE 37.6 Structure of sphingolipids. The nucleus is a highly structured organelle. For a long time, it was thought that lipids, which quantitatively are a minor component of the nucleus, only served a structural role and originated in the cytoplasm. It is now recognized that lipids serve other functions including signalling and modulating. Lipid-metabolizing enzymes have been demonstrated to be present within the nucleus. Phospholipids are the predominant class of lipid in the nucleus, with lesser amounts of cholesterol, free fatty acids, diglycerides and sphingolipids. The nucleus is surrounded by the nuclear envelope, which comprises an outer nuclear membrane and an inner nuclear membrane. Cholesterol has a structural role in the outer nuclear membrane, which is continuous with the endoplasmic reticulum; the inner nuclear membrane is associated with the nuclear lamina and chromatin and is deficient in cholesterol. The inner and outer nuclear membranes are joined by the pore membranes at the nuclear pores, which are associated with the nuclear pore complexes that allow passive transfer of molecules < 50 kDa between the cytoplasm and nucleoplasm. The passage of larger molecules is energy dependent and requires a nuclear localization signal. Lipids with very long chain fatty acids are associated with the pore membrane–nuclear pore complexes and appear to be essential for maintaining their function. The lipoproteins are submicroscopic, macromolecular complexes of lipids (cholesterol, triglycerides, phospholipids) and proteins (apolipoproteins, enzymes), held by non-covalent forces. The basic structure of lipoproteins is a hydrophobic core of triglycerides and/or cholesteryl esters surrounded by a layer of amphipathic phospholipids, unesterified cholesterol and proteins (see Fig. 37.7). The hydrophilic surface protects the hydrophobic core from the aqueous environment. Lipoproteins differ in their relative concentrations of protein to lipid and in their constituent lipids and proteins (Table 37.2). The densities of lipoproteins are inversely related to their size. The lipoproteins can be classified on the basis of their size, density or protein composition. The nomenclature of the lipoproteins is based on their density: chylomicrons (< 0.95 g/mL); VLDL (0.95–1.006 g/mL); intermediate density lipoproteins (IDL) (1.006–1.019 g/mL); low density lipoproteins (LDL) (1.019–1.063 g/mL) and high density lipoproteins (HDL) (1.063–1.210 g/mL). The classes are not homogeneous; each represents a continuum of particles of differing size, density and fate and, in the case of VLDL, also of origin. The physicochemical characteristics of the principal lipoproteins are summarized in Table 37.3. FIGURE 37.7 Generic lipoprotein structure. The apolipoprotein (apo) B-containing lipoproteins contain only one molecule of apo B per lipoprotein particle whereas multiple molecules of the other apolipoproteins are present in other lipoprotein particles. Chylomicrons are the largest and most buoyant class of lipoprotein. The major protein component is apo B-48 but they also contain apo A-I, apo A-II and apo A-IV. After secretion, they acquire apo E and apo C from HDL. Chylomicrons are formed in the intestine and are the transport vehicle for dietary fat. The largest chylomicron particles have a diameter of over 1000 nm, whereas the smallest (75–200 nm) overlap with the apo B-100-containing lipoproteins. Some of the smaller particles within the chylomicron range are this size when they are secreted by enterocytes, while others represent partially delipidated ‘remnant’ particles. The core of chylomicrons is composed predominantly of triglycerides derived from the diet. These are the largest of the lipoproteins containing endogenously produced lipids. The major protein component of VLDL is apo B-100 but they also contain apo C-I, apo C-II, apo C-III, apo E and small amounts of apo A. Like chylomicrons, VLDLs acquire the majority of their component apo E and apo C from HDL in the circulation; the core of VLDLs is composed predominantly of triglycerides. In contrast to chylomicrons, the triglycerides in VLDL are endogenous in origin. These particles are produced during the conversion of VLDL to LDL; their densities lie between those of these lipoproteins. The core of IDLs contains cholesteryl esters and triglycerides. These are the major cholesterol-containing lipoproteins and represent the end-product of VLDL catabolism. The core of LDLs comprises mainly cholesteryl esters; the protein component is apo B-100. These are the smallest and densest of the lipoproteins. They may be sub-classified on the basis of size, density, shape, surface charge and electrophoretic mobility, as well as apolipoprotein composition (Table 37.3). High density lipoprotein is usually divided into three major subclasses. Nascent, discoidal or pre-β1HDL comprises predominantly apo A-I and phospholipid. It is the preferred substrate for the ATP binding cassette transporter A1 (ABCA1), which actively exports free cholesterol from peripheral cells and macrophages. HDL3 is formed from pre-β1HDL by the acquisition of free cholesterol. It is the preferred substrate for lecithin cholesterol acyl transferase (LCAT), which esterifies free cholesterol, increasing the size of the particle and allowing the uptake of more free cholesterol, producing the larger and more cholesterol-rich HDL2. High density lipoprotein particles may contain apo A-I alone (LpA-I), both apo A-I and apo A-II (LpA-I/A-II) or apo A-II alone (LpA-II). LpA-I predominates in HDL2, whereas LpA-I/A-II predominate in HDL3. LpA-II represents a very small proportion of both HDL2 and HDL3. Lipoprotein(a) (Lp(a)) consists of LDL with its apo B-100 bound by a disulfide bond to apolipoprotein(a) (apo(a)). It is thought to be assembled extracellularly, either in the circulation or on the surfaces of hepatocytes. The plasma concentration of Lp(a) is genetically determined and is inversely related to the length of the apo(a), so that the greater the chain length, the lower the concentration. Epidemiological studies suggest that a high Lp(a) concentration is an independent risk factor for cardiovascular disease (CVD), particularly in subjects with familial hypercholesterolaemia. Lipoprotein X is a lipoprotein that is found only in the plasma of subjects with cholestasis or who have familial lecithin cholesterol acyltransferase deficiency. It is composed of phospholipids, free cholesterol and proteins; the major protein is albumin but small amounts of apo C and apo D are also present. It contains no apo B. Unlike all other lipoproteins, it migrates towards the cathode on agarose gel electrophoresis. The apolipoproteins are amphipathic; their hydrophobic regions interact with the lipids in the lipoprotein particle while their hydrophilic regions allow interaction with the aqueous environment. They have three functions: they provide the structural element to the lipoprotein particles, they act as ligands for specific receptors and they also act as activators or inhibitors of specific enzymes involved in lipoprotein metabolism. On the basis of electrophoretic mobility, HDL and LDL were originally referred to as α- and β-lipoproteins. The nomenclature of the corresponding apolipoproteins has arisen from this, apo A being the apolipoprotein derived from HDL (α-lipoprotein) and apo B being derived from LDL (β-lipoprotein). Apolipoprotein A-I (apo A-I) (molecular weight 29 kDa) is the major protein of HDL, constituting 70–80% of HDL protein. It is synthesized primarily in the liver and small intestine. In addition to its structural role in HDL, it is also an activator of lecithin cholesterol acyltransferase (LCAT). Reverse cholesterol transport is dependent on the ability of apo A-I to promote cellular cholesterol efflux, to bind to lipids, to activate LCAT and, within mature HDL, to interact with lipid transfer proteins and specific receptors. The gene for apo A-I (APOA1) is part of a gene cluster on the long arm of chromosome 11 that includes APOC3, APOA4 and APOA5. Epidemiological studies have shown that plasma apo A-I concentrations, like those of HDL-cholesterol (HDL-C), are inversely related to cardiovascular risk. Apolipoprotein A-II (apo A-II) (molecular weight 17 kDa, as homodimer) is also synthesized in the liver and, to a lesser extent, the small intestine. It accounts for about 20% of HDL protein. Some HDL contains apo A-I and apo A-II while some HDL contains apo A-I alone. A small amount of plasma apo A-II is associated with chylomicrons and VLDL. Apo A-II regulates lipoprotein lipase (LPL) activity, and is a cofactor for LCAT and for cholesteryl ester transfer protein (CETP). Like apo A-I, it appears to be inversely related to the risk of coronary disease. It may play a role in the remodelling of HDL, possibly by an effect on the reactivity of HDL towards lipid transfer proteins, enzymes and receptors, including the scavenger receptor B1 (SRB1) (see p. 719). Apolipoprotein A-IV (apo A-IV) (molecular weight 44 kDa) is synthesized only in the small intestine. It has been suggested that it may have a role in intestinal lipid transport, increasing the residence time of nascent chylomicron particles, allowing for greater expansion of their cores and thus capacity to transport triglycerides. The majority of apo A-IV in plasma exists in the free form. A small amount is associated with HDL and chylomicrons. In vitro, apo A-IV activates LCAT, although not as effectively as does apo A-I. It may also be necessary for the maximal activation of LPL by apo C-II. Over-expression of APOA4 in mice results in increased plasma concentrations of total- and HDL-cholesterol, and triglycerides; despite this, it protects against diet-induced atherosclerosis. In humans, apo A-IV deficiency has been reported in patients with apo A-I and apo C-III deficiency, and this may account for the fat malabsorption seen in affected individuals. The APOA5 gene is expressed in liver, and apolipoprotein A-V (apo A-V), in contrast to other apolipoproteins, is present at very low concentrations in the plasma (approximately 5 nmol/L). It is found primarily in HDL. Apo A-V affects plasma triglycerides through an effect on the lipolysis of the triglyceride-rich lipoproteins, possibly by binding to the lipoprotein, endothelial proteoglycans and LPL and thus stabilizing the lipolytic machinery. Although plasma concentrations of apo A-V show little correlation with plasma triglyceride concentration or with prevalence of cardiovascular disease, genetic studies have shown polymorphisms in APOA5 to be strong determinants of both. Genetic variants in humans have been identified, in association with both high and low triglyceride concentrations. Deficiency leads to reduced LPL activity and a type V dyslipidaemia (p. 726). This lipoprotein exists in two forms; apolipoprotein B-100 (apo B-100), which is made in the liver and is the structural protein of VLDL, IDL and LDL, and apo B-48, which is synthesized in the intestine and is incorporated into chylomicrons. Both apo B molecules remain with the lipoprotein particle in which they are secreted throughout the lifespan of that particle, unlike the other apolipoproteins, which readily transfer between different classes of lipoproteins. Increased plasma concentrations of apo B-containing lipoproteins confer an increased risk for the development of atheroma. Both forms of apo B are produced from the APOB gene; post-transcriptional editing of the mRNA in the intestine leads to the production of apo B-48. Apolipoprotein B-100 (apo B-100) (molecular weight 500 kDa) is necessary for the assembly and secretion of VLDL. It contains several very hydrophobic areas that serve as strong lipid-binding domains. It also has several domains that could serve as binding sites for heparin-like molecules and form the basis for some of the cell surface interactions of the apo B-containing lipoproteins. In addition, apo B-100 contains an LDL receptor binding domain (amino acids 3100–3400), which allows the specific uptake of LDL by the LDL receptor. The amino terminal 48% of apo B-100 forms apolipoprotein B-48 (apo B-48) (molecular weight 240 kDa). This apolipoprotein is produced from the APOB gene in the intestine by an mRNA editing process; a cytidine deaminase, APOB mRNA editing enzyme complex 1 (apobec-1), binds to and acts on the cytosine molecule at base 6666 of the mRNA to form a uracil. The editing enzyme complex is only found in intestinal epithelial cells. Its action results in the glutamine 2153 triplet of CAA being converted into the stop codon, UAA. Thus protein synthesis is prematurely terminated at amino acid 2152 and, as a result, apo B-48 does not contain the LDL receptor binding domain present in apo B-100. There are three apolipoprotein Cs, all of which are synthesized in the liver. In plasma, they transfer between the triglyceride-rich lipoproteins (chylomicrons, VLDL and their remnants) and HDL. Apolipoprotein C-I (apo C-I) (molecular weight 7 kDa) forms a minor component of VLDL, IDL and HDL; it acts as an activator of LCAT. Apolipoprotein C-II (apo C-II) (molecular weight 9 kDa) is a component of chylomicrons and VLDL, in which it functions as an activator of LPL. Apo C-II is also found in IDL and HDL. Apolipoprotein C-III (apo C-III) (molecular weight 9 kDa) is synthesized mainly in the liver and, to a lesser extent, the intestine. It forms a major structural component of VLDL but is also present in chylomicrons and HDL. It acts as an inhibitor of LPL, and has more recently been shown to promote hepatic assembly and secretion of VLDL. Apo C-III also inhibits hepatic uptake of chylomicron and VLDL remnant particles, possibly by preventing interaction of apo E on these remnant particles with the hepatic receptor. High plasma apo C-III concentrations are associated with high plasma triglyceride concentrations. Null mutations have been reported which are associated with low plasma triglyceride and LDL, and high HDL concentrations. However, the APOC3 gene is close to the APOA1 gene, and both are deficient in some forms of apo A1 deficiency, which cause low plasma HDL and triglyceride concentrations. Apolipoprotein D (apo D) (molecular weight 33 kDa) is a lipoprotein-associated glycoprotein, forming a minor component of HDL, VLDL, IDL and LDL. It transports small hydrophobic ligands, including sterols and cholesterol. Apo D is associated with increased activity of lipoprotein lipase, and missense mutations cause elevated triglycerides. The apo D concentrations in the hippocampus and cerebrospinal fluid (CSF) of patients with Alzheimer disease are increased. Apolipoprotein E (apo E) is a 299 amino acid glycoprotein (molecular weight 34 kDa) synthesized by the liver and found in all classes of lipoproteins except LDL. It is involved in the control of chylomicron and VLDL remnant removal from the circulation. It also has antioxidant properties, and controls the efflux of cholesterol from cells, together with apo A-I. Apo E is a polymorphic protein: three common isoforms occur, which can be separated by isoelectric focusing and are designated apo E2, apo E3 and apo E4. Apo E2 differs from apo E3 by only a single amino acid, Cys being substituted for Arg at residue 158. Apo E4 also differs from apo E3 by only a single amino acid, Arg being substituted for Cys at residue 112. The apo E3/E3 phenotype is the most common, comprising 50–70% of the population, whereas the apo E2/E2 phenotype is the least common, occurring in about 1% of the population. The apo E3 isoform is associated with normal chylomicron and VLDL metabolism. The E2 isoform does not function as an effective ligand for the receptor-mediated uptake of remnant particles, having less than 2% of normal apo E3 binding to the LDL receptor. As a result, remnant lipoproteins tend to accumulate in the plasma of individuals homozygous for apo E2/E2. The E4 isoform is associated with higher concentrations of LDL-cholesterol than E3. Apo E4 homozygotes are also at increased risk of Alzheimer disease. Apo E synthesis is upregulated to aid cell repair in response to cellular stress or injury. Apo E4 is more susceptible to proteolytic cleavage than E2 or E3. This results in accumulation of intracellular fragments that cause changes in the cytoskeleton and the formation of neurofibrillary tangles. Apolipoprotein M (apo M) (molecular weight 26 kDa) fulfills the criteria for being an apolipoprotein as it is not found free in plasma but is predominantly associated with HDL. Like apo D, it is a member of the lipocalin family of proteins, which contain a binding domain for small lipophilic ligands, and may therefore have a role in transport of small lipid molecules. Although apo M is found in association with only 5% of HDL particles, where it may potentiate the antioxidant effect of HDL, its concentration is positively correlated with cholesterol concentration, suggesting that it may also have a role in cholesterol metabolism. This apolipoprotein (apo(a)) is a large glycated protein of variable size (molecular weight 200–800 kDa). It contains multiple kinks in the polypeptide chain that are termed ‘kringles’. Apo(a) is a homologue of plasminogen; it contains a single copy of plasminogen kringle 5, multiple copies of plasminogen kringle 4 and an inactive protease domain. Kringle 4 shows wide variation in the number of repeats within the apo(a) molecule. In a subpopulation of LDL particles, apo(a) forms disulphide bridges with apo B-100 to form a distinct lipoprotein class termed lipoprotein(a) (Lp(a)) (see p. 708). The function of apo(a) is unknown. It has a strong homology with plasminogen and may interfere with fibrinolysis. Cholesterol of both dietary and biliary origin is absorbed in the upper jejunum facilitated by a specific transporter protein, the Niemann–Pick C1-like 1 (NPC1L1) protein. This has 50% homology with the product of the NPC1 gene, the Niemann–Pick C1 disease protein, which is involved in intracellular cholesterol trafficking. Niemann–Pick C1-like 1 protein transports plant sterols as well as cholesterol. In small intestinal enterocytes, dietary cholesterol is released by the action of lysosomes and selectively esterified by acyl-CoA cholesteryl acyltransferase 2 (ACAT2) prior to incorporation into chylomicrons. Any excess free cholesterol in enterocytes, together with any absorbed plant sterol, is excreted back into the intestinal lumen. The apical excretion of cholesterol and other sterols back into the gut lumen is a function of two hemitransporters of the ATP binding cassette transporter family, ABCG5 and ABCG8. These are also responsible for the excretion of cholesterol and other sterols into the bile by hepatocytes. Expression of ABCG5 and ABCG8 in hepatocytes and enterocytes is under the control of the liver X receptor (LXR) and is induced by cholesterol feeding. Cholesterol absorption reflects the imbalance between the movement of cholesterol across the brush border into enterocytes and its excretion by enterocytes back into the intestinal lumen. In normal individuals, the rate of cholesterol absorption correlates with the plasma sterol concentrations. The rate of cholesterol absorption is a predictor of benefit of statin treatment and response to ezetimibe (a selective inhibitor of NPC1L1). Those with higher rates of cholesterol absorption show reduced CVD benefit from statins, and increased cholesterol lowering with ezetimibe. also referred to as phytosterolaemia, is an autosomal recessive disorder caused by mutations in ABCG5 or ABCG8, in which there is increased absorption of dietary non-cholesterol sterols, and consequently an increased concentration (30–100-fold) of these sterols in blood. A ‘normal’ Western diet contains similar amounts (200–500 mg/day) of cholesterol and of non-cholesterol sterols (mainly plant but also some fish sterols). Approximately 55% of the cholesterol is usually absorbed, but normally less than 1% of the non-cholesterol sterols. In the absence of a functional ABCG5/8 heterodimer, subjects overabsorb cholesterol and non-cholesterol sterols from enterocytes, because these transporters usually excrete some cholesterol and almost all non-cholesterol sterols back into the gut lumen. They also fail to excrete cholesterol and non-cholesterol sterols into the bile. These sterols become deposited at various sites in the body causing tendon xanthomas and premature atherosclerosis. Clinical features also include arthritis and haemolytic episodes. Treatment comprises a diet low in dietary sterols. Ezetimibe is also effective by inhibiting sterol absorption via NPC1L1. Fat (triglyceride) digestion starts in the stomach, where lingual and gastric lipases hydrolyse 25–30% of ingested triglycerides into diglycerides and free fatty acids. In the duodenum, partially digested lipids mix with bile and pancreatic secretions. The latter contains a mixture of enzymes, including carboxyl ester lipase and pancreatic triglyceride lipase, which are capable of further hydrolysing dietary lipids to monoglycerides, free fatty acids, glycerol and cholesterol. The bile acids serve to solubilize the lipids and result in the formation of micelles, the contents of which are absorbed by enterocytes. The absorption of monoglycerides is by passive diffusion. Within the enterocytes, triglycerides are resynthesized from free fatty acids and either monoglycerides or glycerol. Approximately 500 mg of cholesterol is converted, in the liver, to bile acids each day. This replaces the bile acids lost in the stools and represents approximately 5% of the bile acid pool, as the enterohepatic circulation is 95% efficient. The synthesis and secretion of bile acids, together with hepatic cholesterol secretion into bile, represents the major pathway for the elimination of cholesterol from the body. The synthesis of the full complement of bile acids from cholesterol requires 17 enzymatic steps. It is under negative feedback control: accumulation of bile acids leads to reduced activity of the key enzymes 7α-hydroxylase and sterol 12α-hydroxylase. The products of the bile acid synthetic pathway are the primary bile acids, cholic and chenodeoxycholic acids. In the gut, these primary bile acids are converted into secondary and tertiary bile acids by the action of anaerobic bacteria. The bile acids present in bile are a mixture of primary, secondary and tertiary, the latter being a consequence of enterohepatic circulation. A number of enzymatic defects in the synthetic pathway of bile acids have been described; in general, the earlier in the pathway, the earlier in life clinical problems become manifest and the greater their severity. One of these disorders, due to defective sterol 27-hydroxylase activity, is cerebrotendinous xanthomatosis (CTX). In this condition, cholesterol and its 5α-reduced derivative, cholestanol, accumulate in the blood and tissues. As a result of this accumulation, affected individuals develop tendon xanthomas like those found in familial hypercholesterolaemia. In CTX, however, these sterols also accumulate in myelin sheaths, leading to progressive neurological dysfunction. The bile acids act to solubilize fats during absorption from the gut, but they also have a solubilizing action in the bile. If the proportions of bile acids, cholesterol and phospholipid in the bile are disturbed, there is an increased risk of gallstones being formed. Lipoprotein metabolism is summarized in Figure 37.8. FIGURE 37.8 Outline of the metabolism of the apo B-containing lipoproteins. The assembly of the apo B-containing lipoproteins requires the coordinated synthesis of apo B and lipids. First, apo B has lipid added to it by microsomal triglyceride transfer protein (MTP) as it is being synthesized in the endoplasmic reticulum, to form a nascent lipoprotein particle. Further triglyceride may be added, forming VLDL2. In the next step, VLDL2 is exported from the endoplasmic reticulum by a membrane associated protein complex comprising a coatomere protein (COPII) and a GTPase (sar1b). The resulting sar1/COPII vesicles then fuse with the Golgi apparatus. Apo A-IV, apo C-III and apo A-I are added to the surface of the lipoprotein. In the presence of large amounts of free fatty acids and triglycerides, phospholipase D1 and extracellular signal-regulated kinase 2 (ERK2) increase the formation of lipid droplets that deliver lipids to the Golgi apparatus and promote the further addition of lipid to VLDL2, which is thereby converted to VLDL1. In healthy individuals, most VLDL particles are small and relatively triglyceride-poor VLDL2. In conditions such as insulin resistance and type 2 diabetes there is increased production of larger, triglyceride-rich VLDL1, which in turn generate atherogenic remnants, small dense LDL particles and triglyceride rich HDL particles that are more readily catabolized, leading to low concentrations of circulating HDL. Apolipoprotein B is constitutively synthesized, but the rate of production of apo B-containing lipoproteins is related to the availability of fatty acids for triglyceride synthesis. When there is a plentiful supply of fatty acids, the majority of the apo B that is synthesized is incorporated into lipoproteins; however, when fatty acids are in short supply, the apo B is degraded intracellularly. Once lipoprotein particles are fully formed, they are transferred to the cell surface for secretion. Very low density lipoprotein particles (200 nm) and chylomicrons (1000 nm) are large in the context of classic transport vesicles (range 50–80 nm). The intracellular transport of chylomicrons and some VLDL particles relies on sar1/COPII vesicles. Defects in the SARA2 gene that codes for sar1b result in chylomicron retention disease, in which there is an inability to secrete chylomicron particles. Apo B-48 is absent from the circulation, but apo B-100 containing lipoproteins are still present, although in reduced amounts, as sar1b is also involved in later stages of the VLDL pathway. Newly synthesized VLDL is not necessarily secreted from the cell, and may undergo immediate degradation. Sortilins (see p. 721) are now recognized to be involved in routing newly synthesized lipoproteins either to intracellular degradation or for export into the circulation. Enterocytes absorb dietary cholesterol and triglyceride from the gut in the form of free cholesterol, fatty acids and monoglycerides. After re-esterification, cholesteryl esters and triglycerides (containing fatty acids of chain length C14 and greater) are incorporated into the cores of chylomicron particles. Enterocytes synthesize apo B-48 (the major protein of chylomicrons), apo A-I, apo A-II and apo A-IV that, together with phospholipids, form the surface layer of chylomicrons. Apolipoprotein B-48 is essential for chylomicron secretion. Secretory vesicles bud off from the Golgi apparatus and migrate to the basolateral regions of enterocytes. Here, they fuse with the plasma membrane and release chylomicrons into the intestinal lymphatics. The chylomicrons pass in the lymphatics to the thoracic duct and enter the circulation via the left subclavian vein. There is only one apo B-48 molecule per chylomicron particle and it remains with the particle throughout its life until, as a chylomicron remnant, it is taken up by the liver. In contrast, multiple copies of the other apolipoproteins are present in a single chylomicron particle. These other apolipoproteins do not remain with a given chylomicron particle throughout its life but are exchanged with other lipoproteins. From the time of secretion, chylomicrons undergo constant modification, gaining apo C-II, apo C-III, apo E, phospholipids and cholesterol from HDL. The acquisition of apo C-II allows chylomicrons to interact with LPL, which is sited on the vascular endothelium, especially in adipose tissue and muscle. Lipoprotein lipase acts extracellularly to hydrolyse triglycerides within the chylomicron cores; the fatty acids thus released can be either utilized as an energy source or re-esterified and stored in adipose tissue as triglycerides. As hydrolysis proceeds, the cores of the chylomicrons reduce in size and excess surface components – phospholipids, free cholesterol, apo C-II and apo C-III – are transferred back to HDL. Apolipoprotein C-III may exert a modulating effect on LPL-catalysed chylomicron hydrolysis. Both apo C-II and LPL are necessary for normal chylomicron catabolism. Continuing loss of apo C-II as the mass of individual chylomicron particles is reduced eventually prevents further interaction with LPL, and chylomicron remnant particles are generated. These remnants have a relatively high content of cholesteryl esters and apo E. Chylomicron remnants are normally cleared quickly from the plasma by the liver. They enter the space of Disse through the fenestrated sinusoidal endothelium, together with lipoprotein lipase. The space of Disse is rich in heparan sulfate proteoglycans (HSPRG) as well as hepatic lipase (HL) and apo E, which are secreted by hepatocytes. The proteoglycans and apo E bind the remnants that are further catabolized by HL and LPL, becoming enriched in apo E and further depleted in triglycerides. The remnants may then be taken up in a process directly mediated by HSPRG or via apo E binding to the LDL receptor related protein (LRP). This LRP mediated uptake may be of remnant particles alone, or of remnants bound to HSPRG. The LDL receptor is also able to uptake chylomicron remnants via apo E binding in a process independent of HSPRG. The cholesteryl esters delivered to the liver by chylomicron remnant particles may be utilized for the synthesis of bile acids or membranes or be secreted in VLDL, while the apo B-48 undergoes degradation. In summary, chylomicron metabolism essentially comprises two steps. In the first step, most of the fatty acids derived by peripheral lipolysis of chylomicrons enter adipocytes for storage as triglycerides or other cells for oxidation for energy production. A small fraction of the released fatty acids is bound to plasma albumin and transported in the blood to the liver and other tissues. The second step involves remnant particles delivering the remaining triglycerides and almost all the cholesterol to the liver. Triglycerides in white adipose tissue, far from being an inert store, are continuously undergoing lipolysis and re-esterification. In the fasting state, and at times of increased energy demand, free fatty acids are released into the circulation and transported to other tissues. The lipolysis of triglycerides in white adipose tissue is initiated by adipose triglyceride lipase (ATGL). Its action results in the production of diglycerides and free fatty acids, but it has very little hydrolytic activity towards diglycerides. Hormone sensitive lipase, which until recently was thought to be the rate-limiting enzyme involved in the release of free fatty acids from adipose tissue, is actually rate limiting for the hydrolysis of diglycerides rather than triglycerides. The monoglycerides resulting from the action of HSL are acted on by a third enzyme, monoglyceride lipase (MGL). Adipocyte lipolysis is controlled by a number of lipolytic and antilipolytic hormones, including catecholamines and insulin. Hepatocytes are the originators and often also the acceptors of particles involved in the endogenous pathway, which shows many points of similarity with the exogenous pathway. The liver secretes VLDL, a triglyceride-rich lipoprotein. The triglycerides are produced either de novo by hepatocytes or taken up from the plasma. These triglycerides, together with cholesterol derived from chylomicron remnants or from de novo synthesis, are secreted with phospholipids and apo B-100 as nascent VLDL. Some apo C-I, apo C-II, apo C-III and apo E are also present in the nascent VLDL particle, but the majority of these apolipoproteins are probably acquired from HDL within the circulation in the same way as in the exogenous pathway. In situations where there is an excess of hepatic triglyceride, large VLDL particles are secreted. Large VLDL particles are also secreted in familial hypertriglyceridaemia, whereas in familial combined hyperlipidaemia (see p. 724), the rate of VLDL secretion is increased but a relative scarcity of triglycerides ensures that the individual VLDL particles are smaller and relatively poor in triglycerides. The initial metabolic transformation of VLDL is a progressive LPL-mediated lipolysis analogous to the process involving chylomicrons. This requires apo C-II and produces cholesteryl esters and apo E-rich remnant particles. As in chylomicron metabolism, the surface components of VLDL are transferred back to HDL as the cores shrink. VLDL remnants comprise small VLDL particles and IDL. About half are cleared by the liver in a process that involves uptake by LRP, which recognizes apo E. The remainder undergoes further hydrolysis by HL to form LDL. Low density lipoprotein is the major cholesterol-carrying lipoprotein in the plasma and usually accounts for 70% or more of the total plasma cholesterol. Virtually the only protein contained in the LDL particle is a single molecule of apo B-100, which acts as the ligand for the LDL receptor. LDL receptors are present on hepatocytes as well as the cells of peripheral tissues. Approximately 50% of plasma LDL uptake by the LDL receptor-mediated mechanism is hepatic. The major determinant of plasma LDL-C concentration is the number of functional LDL receptors. Low density lipoprotein receptors recognize both apo B-100 on LDL and apo E on remnant particles and HDL. Once a lipoprotein has been bound to the receptor, the receptor-lipoprotein complex localizes in the coated pit region from where it is internalized by endocytosis. The LDL receptor is recycled while the lipoprotein undergoes lysosomal degradation to unesterified cholesterol and amino acids. The cholesterol thereby released is available for further metabolic transformations as well as to regulate the transcription and/or translation of the HMG-CoA reductase and the LDL receptor genes. The cholesterol may be re-esterified by the action of ACAT and stored, or may be utilized for bile acid, steroid or membrane synthesis. The liver is the key organ for the regulation of cholesterol; not only is it responsible for the majority of cholesterol synthesis but it also acquires cholesterol from all lipoprotein classes. The cholesterol secreted into bile is mostly derived from lipoproteins with only a small contribution coming from de novo hepatic synthesis or hepatic cholesteryl ester stores. This involves preferential trafficking of lipoprotein-derived cholesterol, which involves multiple cholesterol transport-related gene products, the expression of which is regulated by the concerted activity of sterol-activated transcription factors. FIGURE 37.9 Outline of HDL metabolism and its role in reverse cholesterol transport. Apo A-I is exported into the circulation by liver and intestinal cells. Here it may combine with phospholipids to form nascent (discoidal or pre-β1) HDL. Pre-β1
Lipids and disorders of lipoprotein metabolism
INTRODUCTION
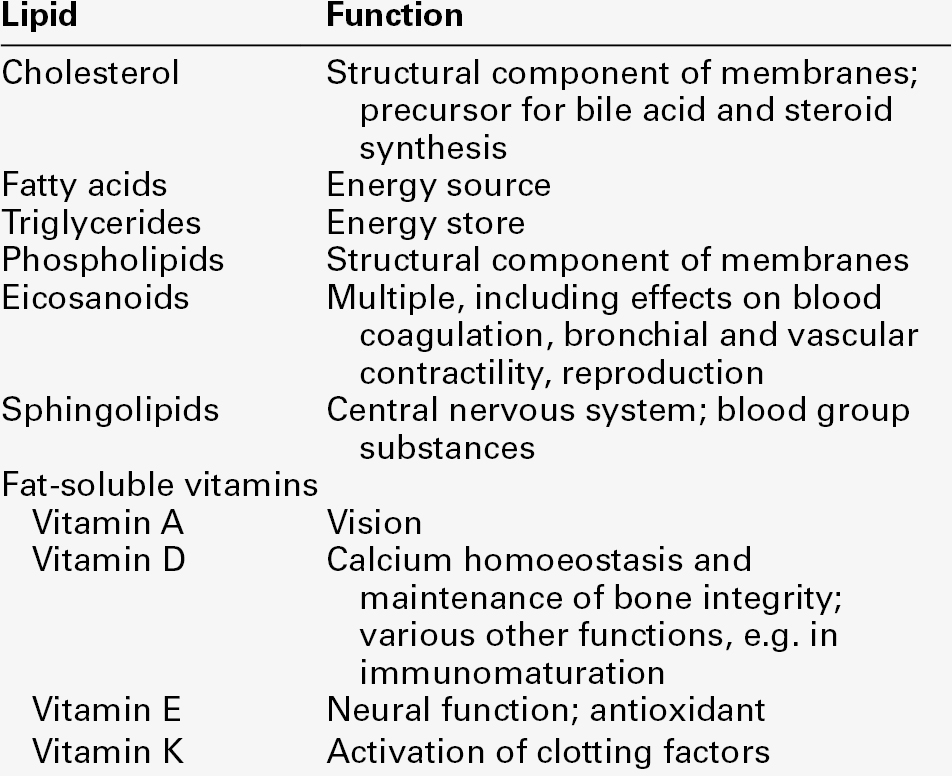
LIPIDS
Sterols
Cholesterol
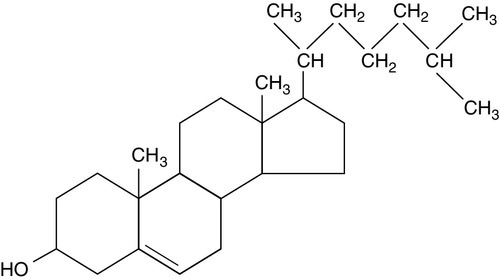
Cholesterol and membranes
Phytosterols
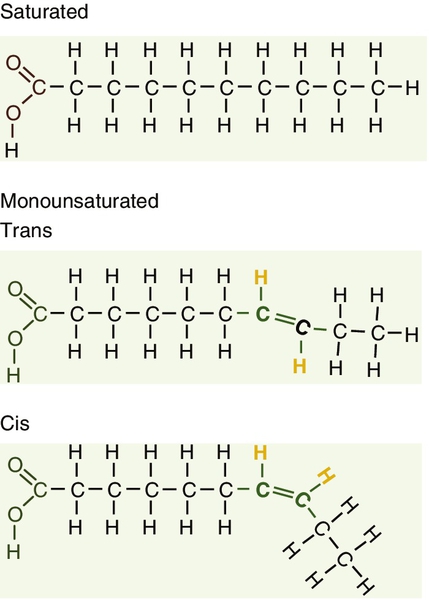
Fatty acids
Triglycerides
Phospholipids
Eicosanoids
Sphingolipids
Nuclear lipids
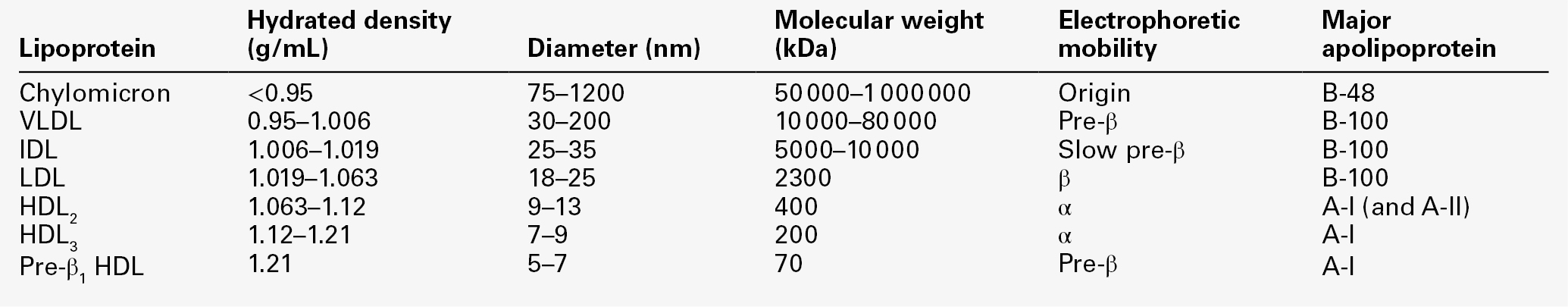
LIPOPROTEINS
Chylomicrons
Very low density lipoproteins
Intermediate density lipoproteins
Low density lipoproteins
High density lipoproteins
Lipoprotein(a)
Lipoprotein X
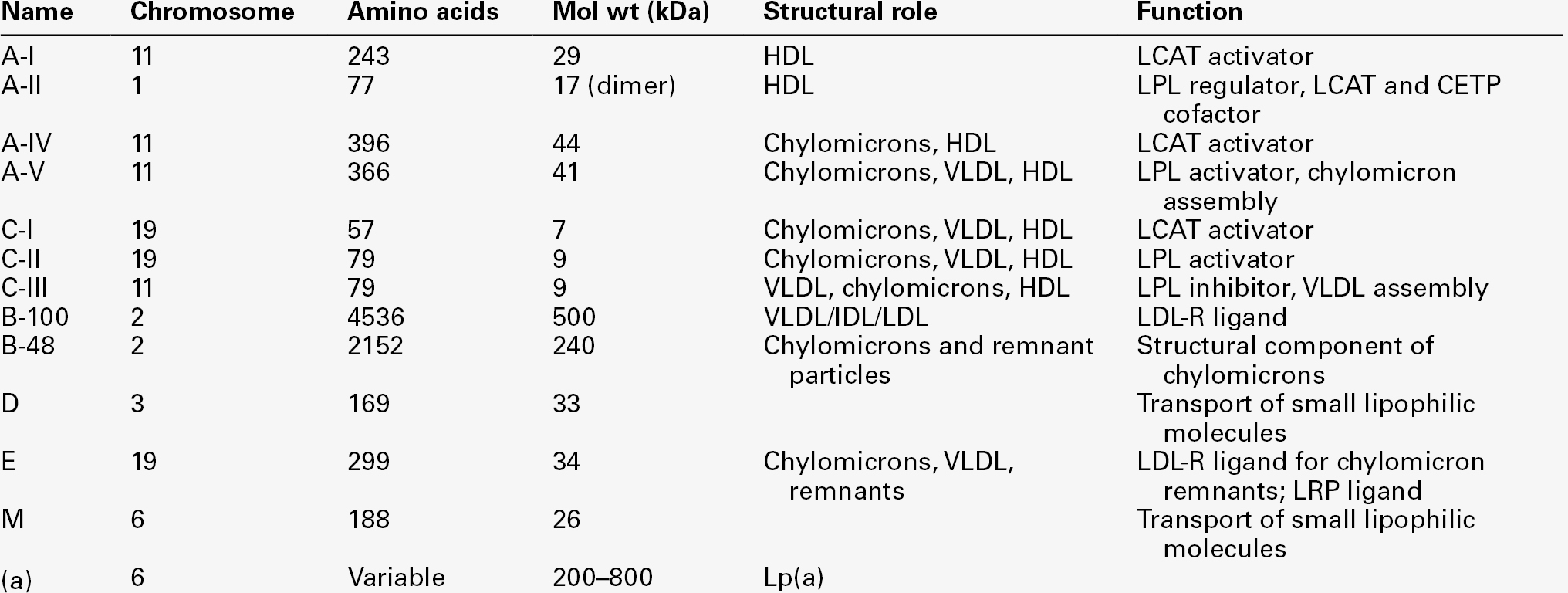
APOLIPOPROTEINS
Apolipoprotein A
Apolipoprotein A-I
Apolipoprotein A-II
Apolipoprotein A-IV
Apolipoprotein A-V
Apolipoprotein B
Apolipoprotein B-100
Apolipoprotein B-48
Apolipoprotein C
Apolipoprotein C-I
Apolipoprotein C-II
Apolipoprotein C-III
Apolipoprotein D
Apolipoprotein E
Apolipoprotein M
Apolipoprotein(a)
CHOLESTEROL ABSORPTION
β-Sitosterolaemia
TRIGLYCERIDE DIGESTION
BILE ACID METABOLISM
LIPOPROTEIN METABOLISM
Assembly of apolipoprotein B-containing lipoproteins
Exogenous pathway
Lipolysis in adipose tissue
Endogenous pathway
Hepatic cholesterol trafficking
High density lipoprotein metabolism (see Fig. 37.9)
Assembly of lipoproteins
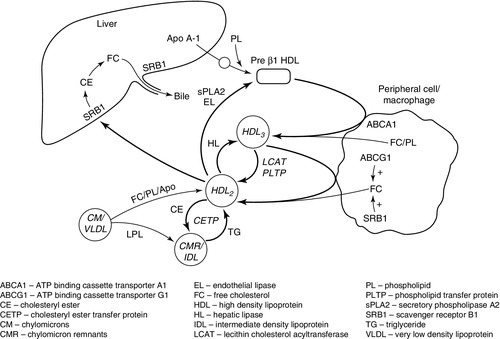
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


