CHAPTER OUTLINE
Histone Modifications and Chromatin Structure
Checkpoints and Cell Cycle Regulation
Tumor Suppressors and Cell Cycle Regulation
Additional Activities of DNA Polymerases
Telomere Replication: Implications for Aging and Disease
Trinucleotide Repeat Expansion
Postreplicative Modification of DNA: Methylation
DNA Methylation and Epigenetics
DNA Methylation and Imprinting
High-Yield Terms
Phosphodiester bond: chemical bond between the 5’-phosphate of one base and the 3’-hydroxyl of an adjacent base in a nucleic acid
Watson-Crick base-pair: refers to the normal hydrogen bonding that occurs in a duplex of DNA, or an RNA-DNA duplex, where A bonds to T and G bonds to C
Annealing: with respect to nucleic acids this refers to DNA or RNA pairing by hydrogen bonds to a complementary sequence, forming a double-stranded polynucleotide
Nucleosome: a structure formed by DNA and protein interaction that consisting of an octamer protein structure composed of 2 subunits each of histones H2A, H2B, H3, and H4
Chromatin: the combination of DNA and proteins that make up the contents of the nucleus of a cell
Heterochromatin: densely packed region of a chromosome often found near the centromeres, is generally transcriptionally silent
Euchromatin: loosely packed regions of a chromosome where active gene transcription can occur
Telomere: a region of repetitive nucleotide sequences at each end of a chromatid
Cyclin: any of a number of proteins whose levels fluctuate during the eukaryotic cell cycle; these proteins regulate the activity of a family of kinases involved in controlling passage through the cell cycle
Proof-reading: in the process of DNA replication this refers to the 3′ → 5′ exonuclease activity of DNA polymerase, which allows it to remove an incorrectly incorporated nucleotide, recognized by non–Watson-Crick base-pairing
Okazaki fragment: short stretches of newly synthesized DNA representing the lagging strand
Topoisomerase: enzymes that cleave the DNA strand, thereby relieving torsional stresses incurred during the process of DNA replicaiton
Epigenetics: defines the mechanism by which changes in the pattern of inherited gene expression occur in the absence of alterations or changes in the nucleotide composition of a given gene
Imprinting: refers to the fact that the expression of some genes depends on whether or not they are inherited maternally or paternally
Trinucleotide repeat: sequences of 3 nucleotides repeated in tandem on the same chromosome a number of times, abnormal expansion of the repeat is associated with the genesis of disease
DNA Structure
DNA is a specialized form of polynucleotides. Polynucleotides are formed by the condensation of 2 or more nucleotides. The condensation most commonly occurs between the alcohol of a 5′-phosphate of one nucleotide and the 3′-hydroxyl of a second, with the elimination of H2O, forming a phosphodiester bond. The formation of phosphodiester bonds in DNA and RNA exhibits directionality. The primary structure of DNA and RNA (the linear arrangement of the nucleotides) proceeds in the 5′ → 3′ direction (Figure 35-1). The common representation of the primary structure of DNA or RNA molecules is to write the nucleotide sequences from left to right synonymous with the 5′ → 3′ direction.

FIGURE 35-1: A segment of one strand of a DNA molecule in which the purine and pyrimidine bases guanine (G), cytosine (C), thymine (T), and adenine (A) are held together by a phosphodiester backbone between 2′-deoxyribosyl moieties attached to the nucleobases by an N-glycosidic bond. Note that the backbone has a polarity (ie, a direction). Convention dictates that a single-stranded DNA sequence is written in the 5′ to 3′ direction (ie, pGpCpTpA, where G, C, T, and A represent the 4 bases and p represents the interconnecting phosphates). Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry. 29th, ed. New York: McGraw-Hill; 2012.
Within cells, DNA exists as a helix of 2 complementary antiparallel strands, wound around each other in a rightward direction and stabilized by hydrogen bonding (H-bonds) between nucleobases (bases) in adjacent strands (Figure 35-2). The bases are in the interior of the helix aligned at a nearly 90-degree angle relative to the axis of the helix. Due to the fact that purine bases form H-bonds with pyrimidines, in any given molecule of DNA the concentration of adenine (A) is equal to thymine (T) and the concentration of cytidine (C) is equal to guanine (G). This means that A will only base-pair with T, and C with G. According to this pattern, known as Watson-Crick base-pairing, the base-pairs composed of G and C contain 3 hydrogen bonds, whereas those of A and T contain 2 hydrogen bonds (Figure 35-3). This makes G-C base-pairs more stable than A-T base-pairs.

FIGURE 35-2: A diagrammatic representation of the Watson and Crick model of the double-helical structure of the B form of DNA. The horizontal arrow indicates the width of the double helix (20 Å), and the vertical arrow indicates the distance spanned by one complete turn of the double helix (34 Å). One turn of B-DNA includes 10 base pairs (bp), so the rise is 3.4 Å per bp. The central axis of the double helix is indicated by the vertical rod. The short arrows designate the polarity of the antiparallel strands. The major and minor grooves are depicted. (A, adenine; C, cytosine; G, guanine; P, phosphate; S, sugar [deoxyribose]; T, thymine.) Hydrogen bonds between A/T and G/C bases indicated by short, red, horizontal lines. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.

FIGURE 35-3: DNA base pairing between adenine and thymine involves the formation of 2 hydrogen bonds. Three such bonds form between cytidine and guanine. The broken lines represent hydrogen bonds. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
The antiparallel nature of the helix stems from the orientation of the individual strands. From any fixed position in the helix, one strand is oriented in the 5′ → 3′ direction and the other in the 3′ → 5′ direction. On its exterior surface, the double helix of DNA contains 2 deep grooves between the ribose-phosphate chains. These 2 grooves are of unequal size and termed the major and minor grooves (see Figure 35-2). The difference in their size is due to the asymmetry of the deoxyribose rings and the structurally distinct nature of the upper surface of a base-pair relative to the bottom surface.
The double helix of DNA has been shown to exist in several different forms, depending upon sequence content and ionic conditions of crystal preparation. The B-form of DNA prevails under physiological conditions of low ionic strength and a high degree of hydration. Regions of the helix that are rich in pCpG dinucleotides can exist in a novel left-handed helical conformation termed Z-DNA. This conformation results from a 180-degree change in the orientation of the bases relative to that of the more common A- and B-DNA.
Thermal Properties of DNA
As cells divide, it is a necessity that the DNA be copied (replicated) in such a way that each daughter cell acquires the same amount of genetic material. In order for this process to proceed the 2 strands of the helix must first be separated in a process termed denaturation. DNA denaturation can also be accomplished experimentally by subjecting it to high temperature. Under these conditions the H-bonds between bases become unstable and the strands of the helix separate in a process of thermal denaturation. When thermally melted DNA is cooled, the complementary strands will again re-form the correct base-pairs in a process termed annealing or hybridization. The rate of annealing is dependent upon the nucleotide sequence of the 2 strands of DNA.
Regions of DNA that have predominantly A-T base-pairs will be less thermally stable than those rich in G-C base-pairs. In the process of thermal denaturation, the temperature required for 50% of the DNA molecule to exist single stranded is referred to as the melting temperature (Tm). The Tm is a characteristic of the base composition of that DNA molecule and the conditions of the solution containing the DNA.
Chromatin Structure
Chromatin is a term designating the structure in which DNA exists within cells. The structure of chromatin is determined and stabilized through the interaction of the DNA with DNA-binding proteins. There are 2 classes of DNA-binding proteins. The histones are the major class of DNA-binding proteins involved in maintaining the compacted structure of chromatin. There are 5 different histone proteins identified as H1, H2A, H2B, H3, and H4.
The other class of DNA-binding proteins is a diverse group of proteins called simply, nonhistone proteins. This class of proteins includes the various transcription factors, polymerases, hormone receptors, and other nuclear enzymes. In any given cell there are greater than 1000 different types of nonhistone proteins bound to the DNA.
The binding of DNA by the histones generates a structure called the nucleosome (Figure 35-4). The nucleosome core contains an octamer protein structure consisting of 2 subunits each of H2A, H2B, H3, and H4. Histone H1 occupies the inter-nucleosomal DNA and is identified as the linker histone. The nucleosome core contains approximately 150 bp of DNA. The nucleosomes would appear as “beads-on-a-string” if the DNA were pulled into a linear structure and observed under an electron microscope. The nucleosome cores themselves coil into a solenoid shape which itself coils to further compact the DNA. These final coils are compacted further into the characteristic chromatin structure seen in a metaphase karyotype analysis (Figure 35-5).

FIGURE 35-4: Components of a nucleosome. Nucleosome is a structure that produces the initial organization of free double-stranded DNA into chromatin. Each nucleosome has an octomeric core complex made up of 4 types of histones, 2 copies each of H2A, H2B, H3, and H4. Around this core is wound DNA approximately 150 bp in length. One H1 histone is located outside the DNA on the surface of each nucleosome. DNA associated with nucleosomes in vivo thus resembles a long string of beads. Nucleosomes are very dynamic structures, with H1 loosening and DNA unwrapping at least once every second to allow other proteins, including transcription factors and enzymes, access to the DNA. Mescher AL. Junqueira’s Basic Histology Text and Atlas, 13th ed. New York: McGraw-Hill; 2013.

FIGURE 35-5: Levels of DNA packing in a typical metaphase chromosome (top) down to a naked duplex of DNA (bottom). The “beads-on-a-string” structure, formed as histones form nucleosomes and become wrapped by the DNA, is visible as such under electron microscopy. The DNA-protein complex within the cell is attached to a scaffold composed of highly specialized DNA-binding proteins that aid in the condensation of the chromosome to the metaphase stage. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
In a broad consideration of chromatin structure there are 2 forms: heterochromatin and euchromatin. Heterochromatin is more densely packed than euchromatin and is often found near the centromeres of the chromosomes. Heterochromatin is generally transcriptionally silent. Euchromatin on the other hand is more loosely packed and is where active gene transcription can occur.
Histone Modifications and Chromatin Structure
Histone acetylation is known to result in a more open chromatin structure and these modified histones are found in regions of the chromatin that are transcriptionally active. Proteins are known to interact with acetylated histones that together lead to a more open chromatin structure. Proteins that bind to acetylated lysines in histones contain a domain called a bromodomain. Conversely, underacetylation of histones is associated with closed chromatin and transcriptional inactivity. A direct correlation between histone acetylation and transcriptional activity was demonstrated when it was discovered that protein complexes, previously known to be transcriptional activators, were found to have histone acetylase activity. And as expected, transcriptional repressor complexes were found to contain histone deacetylase activity (Chapter 36).
Another histone modification known to affect chromatin structure is methylation. However, with histone methylation there is not a direct correlation between the modification and a specific effect on transcription. Some histone methylations promote an open chromatin structure and thereby, lead to transcriptional activation, whereas other methylations are known to be associated with transcriptionally inactive genes. Proteins that bind to methylated histones contain a domain called chromodomain. The chromodomain is also found in the RNA-induced transcriptional silencing (RITS) complex which involves small interfering RNA (siRNA) and microRNA (miRNA)-mediated downregulation of transcription (Chapter 36).
There are 2 primary mechanisms operating in a dynamic manner to alter chromatin. These mechanisms are methylation of cytidine residues in specific locations in the DNA (see below) and histone modification. Histone modifications include acetylation, methylation, phosphorylation, and ubiquitination (Table 35-1).

Eukaryotic Cell Cycles
Most eukaryotic cells will proceed through an ordered series of events in which the cell duplicates its contents and then divides into two cells. This cycle of duplication and division is called the cell cycle. In order to maintain the fidelity of the developing organism this process of cell division in multicellular organisms must be highly ordered and tightly regulated. The loss of control will lead to abnormal development and is the cause of cancer. The eukaryotic cell cycle is composed of four steps, or phases identified as G1, S, G2, and M (Table 35-2). Not all cells continue to divide during the life span of an organism. Many cells undergo what is referred to as terminal differentiation and become quiescent and no longer divide and reside in a cell cycle phase termed G0. Under certain conditions, such as that resulting from an external signal stimulating cell growth, cells can exit the quiescent state and reenter the cell cycle.

During M-phase there is an ordered series of events that leads to the alignment and separation of the duplicated chromosomes (called sister chromatids). The steps of mitosis are termed prophase, prometaphase, metaphase, anaphase, and telophase. Although cytokinesis is the process by which the parental cell is physically separated into 2 new daughter cells, it actually begins during anaphase. During prophase the duplicated chromosomes condense while outside the nucleus the mitotic spindle assembles between the 2 centrosomes. The centrosome is an organelle that serves as the main microtubule-organizing center that is involved in the attachment of microtubules to the sister chromatids. During prometaphase the nuclear membrane breaks apart and the chromosomes can attach to spindle microtubules and begin active movement. During metaphase the chromosomes are aligned at the equator of the spindle midway between the spindle poles. The sister chromatids are attached to opposite poles of the spindle. During anaphase the sister chromatids synchronously separate to form the 2 sets of daughter chromosomes. Each sister chromatid is slowly pulled toward the spindle pole it faces. During telophase the 2 daughter chromosomes arrive at the spindle poles and decondense. A new nuclear envelope forms around each set of chromosomes which forms the 2 new nuclei. This process marks the end of mitosis and sets the stage for cytokinesis.
Checkpoints and Cell Cycle Regulation
The processes that drive a cell through the cell cycle must be highly regulated so as to ensure that the resultant daughter cells are viable and each contains the complement of DNA found in the original parental cell. Many important genes involved in the regulation of cell cycle transit have been identified and are referred to as cell division cycle genes (or cdc genes). Many of these genes encode proteins that control progression through the phases of a cell cycle at specific points called checkpoints or restriction points.
In addition to cell cycle checkpoints there is the need for cell cycle control mechanisms to exert their influences at specific times during each transit through a cell cycle. The heart of this timing control is the responsibility of a family of protein kinases that are called cyclin-dependent kinases, CDK (Table 35-3). The kinase activity of these enzymes rises and falls as the cell progresses through a cell cycle. Different CDK operate at different points in the cell cycle. The oscillating changes in the activity of CDK leads to oscillating changes in the phosphorylation of various intracellular proteins. These phosphorylations alter the activity of the modified proteins which then effect changes in events of the cell cycle. The cyclical activity of each CDK is controlled by a complex series of proteins, the most important of which are the cyclins, hence the name of the enzymes as cyclin-dependent kinases. The cyclins are so called because their levels cycle up and down as the cell progresses through the phase of the cell cycle. Four different classes of cyclins have been defined on the basis of the stage of the cell cycle in which they bind and activate CDKs. These 4 classes are G1-cyclins, G1/S-cyclins, S-cyclins, and M-cyclins.

In addition to control of CDK kinase activity by cyclin binding, control is exerted to inhibit CDK activity through interaction with inhibitory proteins as well as by inhibitory phosphorylation events. Thus, there is extremely tight control on the overall activity of each CDK. Proteins that bind to and inhibit cyclin-CDK complexes are called CDK inhibitory proteins (CKI, for cyclin-kinase inhibitor). Mammalian cells express 2 classes of CKI. These are called CIP for CDK inhibitory proteins and INK4 for inhibitors of kinase 4. The CIP bind and inhibit CDK1, CDK2, CDK4, and CDK6 complexes, whereas the INK4 bind and inhibit only the CDK4 and CDK6 complexes. There are at least 3 CIP proteins (p21, p27, and p57) and 4 INK proteins (p15, p16, p18, and p19) in mammalian cells. Working in concert, each of these proteins plays a critical role in controlling entry of cells into the cell cycle in response to mitogenic stimuli and to restrict entry in the presence of DNA damage (Figure 35-6).

FIGURE 35-6: Overview of cell cycle control. Cells enter the cell cycle in response to various growth and mitogenic signals. These signals activate the modification of preexisting regulatory proteins and also induce the expression of genes encoding regulatory proteins. If a cell sustains DNA damage, cell cycle inhibitors can be activated to ensure that the cell does not enter S-phase until the damage has been repaired. Reproduced with permission of themedicalbiochemistrypage, LLC.
The cyclical degradation of the cyclins is effected through the action of several different ubiquitin ligase complexes (Chapter 37). There are 2 important ubiquitin ligase complexes that control the turnover of cyclins and other cell cycle regulating proteins. One is the SCF complex which functions to control the transit from G1 to S-phase and the other is called anaphase-promoting complex (APC), which controls the levels of the M-phase cyclins as well as other regulators of mitosis.
Tumor Suppressors and Cell Cycle Regulation
Tumor suppressors (Chapter 52) are so called because cancer ensues as a result of a loss of their normal function, that is, these proteins suppress the ability of cancer to develop. Several tumor suppressor proteins are required for regulation of cell cycle progression. Following the isolation and characterization of 2 tumor suppressor genes in particular it was found that they function to control the ability of cells to progress beyond important checkpoints in the cell cycle. The protein encoded by the retinoblastoma susceptibility gene (pRB) and the p53 protein are both tumor suppressors. The function of pRB is to prevent cells from exiting G1 (see Figure 52-3) and that of p53 is to inhibit progression from G1 to S-phase as well as from S-phase to M-phase (see Figure 52-4).
DNA Replication
As pointed out above, replication of DNA occurs during S-phase of normal cell division cycles. Because the genetic complement of the resultant daughter cells must be the same as the parental cell, DNA replication must possess a very high degree of fidelity. The entire process of DNA replication is complex and involves multiple enzymatic activities with the actual replication being catalyzed by DNA polymerases (Table 35-4).

Eukaryotic cells express several DNA polymerases that function in distinct types of DNA replication. These DNA polymerases are divided into 4 large families designated A, B, X, and Y (Table 35-5). In addition, there is a reverse transcriptase activity associated with telomerase.

The process of DNA replication begins at specific sites in the chromosomes termed origins of replication, requires a primer bearing a free 3′–OH, proceeds specifically in the 5′ → 3′ direction on both strands of DNA concurrently and results in the copying of the template strands in a semiconservative manner. The semiconservative nature of DNA replication means that the newly synthesized daughter strands remain associated with their respective parental template strands.
At each origin of replication the strands of DNA are dissociated and unwound in order to allow access to DNA polymerase. Unwinding of the duplex at the origin as well as along the strands as the replication process proceeds is carried out by helicases. The resultant regions of single-stranded DNA are stabilized by the binding of single-strand binding proteins. The stabilized single-stranded regions are then accessible to the enzymatic activities required for replication to proceed. The site of the unwound template strands is termed the replication fork (Figure 35-7).
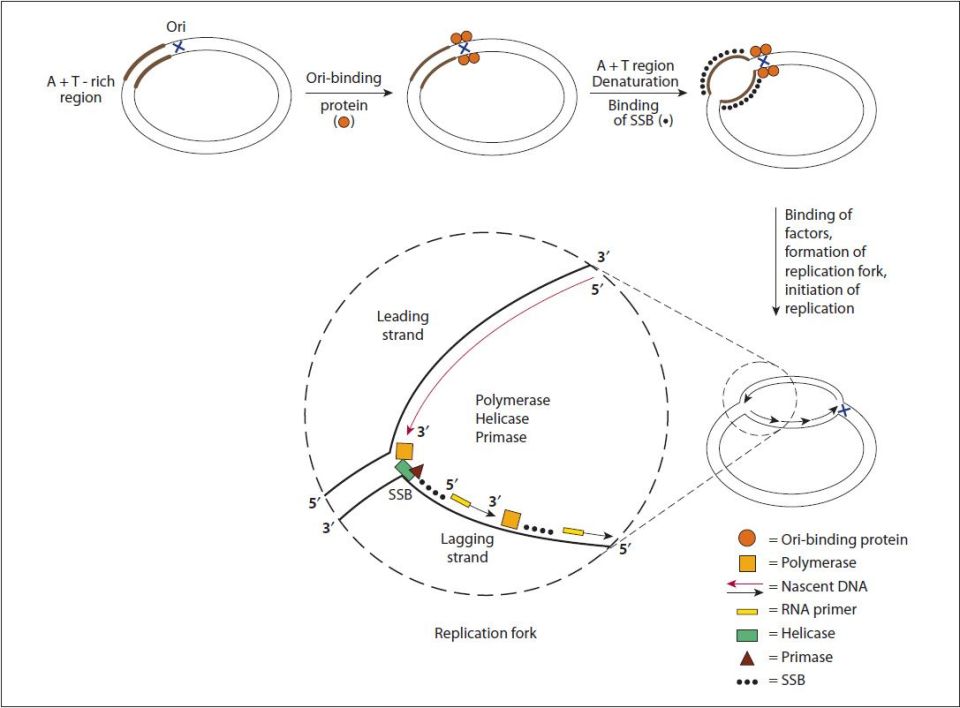
FIGURE 35-7: Steps involved in DNA replication. This figure describes DNA replication in an Escherichia coli cell, but the general steps are similar in eukaryotes. A specific interaction of a protein to the origin of replication (Ori) results in local unwinding of DNA at an adjacent A+T-rich region. The DNA in this area is maintained in the single-strand conformation (ssDNA) by single-strand–binding proteins (SSBs). This allows a variety of proteins, including helicase, primase, and DNA polymerase to bind and to initiate DNA synthesis. The replication fork proceeds as DNA synthesis occurs continuously (long red arrow) on the leading strand and discontinuously (short black arrows) on the lagging strand. The nascent DNA is always synthesized in the 5′ to 3′ direction, as DNA polymerases can add a nucleotide only to the 3′ end of a DNA strand. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York: McGraw-Hill; 2012.
In order for DNA polymerases to synthesize DNA they must encounter a free 3′–OH which is the substrate for attachment of the 5′–phosphate of the incoming nucleotide. During repair of damaged DNA the 3′–OH can arise from the hydrolysis of the backbone of one of the two strands. During replication the 3′–OH is supplied through the use of an RNA primer, synthesized by the primase activity which is DNA polymerase α.
Synthesis of DNA proceeds in the 5′ → 3′ direction through the attachment of the 5′–phosphate of an incoming dNTP to the existing 3′–OH in the elongating DNA strands with the concomitant release of pyrophosphate. While incorporating dNTP into DNA in the 5′ → 3′ direction DNA plymerases move in the 3′ → 5′ direction with respect to the template strand. Synthesis proceeds bidirectionally, with one strand in each direction being copied continuously and one strand in each direction being copied discontinuously. The strand of DNA synthesized continuously is termed the leading strand and the discontinuous strand is termed the lagging strand. The lagging strand of DNA is composed of short stretches of RNA primer plus newly synthesized DNA approximately 100- to 200-base long. The lagging strands of DNA are also called Okazaki fragments. The gaps that exist between the 3′–OH of one leading strand and the 5′–phosphate of another as well as between one Okazaki fragment and another are repaired by DNA ligases, thereby completing the process of replication.
How is it that DNA polymerase can copy both strands of DNA in the 5′ → 3′ direction simultaneously? DNA polymerases exist as dimers associated with the other necessary proteins at the replication fork in a complex identified as the replisome. The template for the lagging strand is temporarily looped through the replisome such that the DNA polymerases are moving along both strands in the 3′ → 5′ direction simultaneously for short distances, the distance of an Okazaki fragment (Figure 35-8).

FIGURE 35-8: Details of the mechanism by which both strands of DNA are replicated simultaneously in the same direction. A portion of the lagging strand is looped around through the DNA polymerase holoenzyme complex such that short stretches of 500–1000 can be continuously replicated in the same direction as the leading strand. Eventually torsional stress will result in disassociation of the enzyme complex and the looping process will need to begin again. This is what results in the average length of the Okasaki fragments generated from the lagging strand. Only DNA helicase, single-strand binding proteins (SSBPs), and the DNA polymerase complex are shown. Reproduced with permission of themedicalbiochemistrypage, LLC.
The progression of the replication fork requires that the DNA ahead of the fork be continuously unwound. Due to the fact that eukaryotic chromosomal DNA is attached to a protein scaffold the progressive movement of the replication fork introduces severe torsional stress into the duplex ahead of the fork. This torsional stress is relieved by DNA topoisomerases. Topoisomerases relieve torsional stresses in duplexes of DNA by introducing either double- (topoisomerases II) or single-stranded (topoisomerases I) breaks into the backbone of the DNA. These breaks allow unwinding of the duplex and removal of the replication-induced torsional strain. The nicks are then resealed by the topoisomerases.
Additional Activities of DNA Polymerases
The main enzymatic activity of DNA polymerases is the 5′ → 3′ synthetic activity. However, DNA polymerases possess 2 additional activities of importance for both replication and repair. These additional activities include a 5′ → 3′ exonuclease function and a 3′ → 5′ exonuclease function. The 5′ → 3′ exonuclease activity allows the removal of ribonucleotides of the RNA primer, utilized to initiate DNA synthesis, along with their simultaneous replacement with deoxyribonucleotides by the 5′ → 3′ polymerase activity. The 5′ → 3′ exonuclease activity is also utilized during the repair of damaged DNA. The 3′ → 5′ exonuclease function is utilized during replication to allow DNA polymerase to remove mismatched bases, termed “proof-reading.” These mismatched bases are recognized by the polymerase immediately due to the lack of Watson-Crick base-pairing.
Telomere Replication: Implications for Aging and Disease
Telomeres are the specialized DNA structures at the ends of all chromosomes. Telomeres consist of repetitive DNA sequences and nucleoproteins, the overall structure of which is referred to as a nucleoprotein cap. The telomere sequence on the lagging strand is composed of the repeat 5′–TTAGGG–3′. The telomeric repeat sequence spans up to several kilobases and is involved in protecting the ends of the chromosomes from exonuclease activity.
The telomeric ends of the lagging strand of each chromosome require a unique method of replication which involves the activity of the enzyme complex called telomerase. Telomerase is complex composed of several proteins, an RNA with sequence complimentary to the telomeric repeats, and a reverse transcriptase activity that extends the 3′-end of the lagging strands using the telomerase RNA as the template (Figure 35-9). The reverse transcriptase activity of telomerase is encoded by the TERT gene (telomerase reverse transcriptase) and the RNA component is encoded by the TERC gene (telomerase RNA component). The TERC RNA contains a repeating hexanucleotide sequence, 3′–AAUCCC–5′, that spans between 3 and 20 kilobases. This sequence in the TERC RNA forms a duplex with the lagging DNA strand at the ends of the chromosomes. The 3′-end of the lagging strand then serves as the primer for the reverse transcriptase activity (TERT) which extends the 3′-end of the chromosome using the TERC RNA as a template. The telomerase process extends the end of the lagging strand that can then be replicated by normal DNA polymerase, thereby preserving the length of the chromosome.

FIGURE 35-9: Steps in telomere replication by telomerase. Reproduced with permission of themedicalbiochemistrypage, LLC.
Telomere shortening is associated with the activation of programmed cell death (apoptosis), loss of tissue stem cells, disease progression, and the overall processes of aging. Decreased telomere length in peripheral blood leukocytes has been shown to correlate with higher mortality rates in older (> 60 years of age) individuals. This is contrasted by studies in centenarians and their offspring that have shown a positive link between telomere length and longevity. Individuals with longer telomeres exhibit an overall healthier profile relative to individuals of similar age with telomeres of shorter length. There is also an intriguing correlation between telomere length and psychological stress and the risk for development of psychiatric disease.
Telomere maintenance correlated to a healthy lifespan is also inferred from studies of various inherited degenerative disorders. For example, individuals harboring a mutation in either the TERT or TERC genes develop autosomal dominant dyskeratosis congenita, DKS (characterized by a triad of abnormal nails, reticular skin pigmentation, and oral leukoplakia). DKS patients have shortened telomeres, a reduced lifespan, and exhibit signs of accelerated ageing. In addition to inherited disorders, telomere shortening is also correlated with acquired degenerative conditions associated with chronically elevated tissue turnover. For example, cirrhosis of the liver is associated with a progressive decline in telomere length.
Trinucleotide Repeat Expansion
The trinucleotide repeat disorders (also known as trinucleotide repeat expansion disorders or triplet repeat expansion disorders) are a set of genetic disorders caused by an increase in the number of trinucleotide repeats in certain genes exceeding the normal, stable, threshold. Each gene affected by trinucleotide repeat expansion has a different number of repeats that constitute the normal threshold and the number that results in manifestation of disease. Why some repeats expand more than others remains an important unresolved question as does the precise mechanism by which the repeats expand. Repeat expansion may occur during normal DNA replication, during replication-dependent repair processes, or during some form of excision repair process, and any one possible mechanism could be operative in different types of repeat expansion disease. Currently, repeat expansion is thought to occur through the formation of looped intermediates which are then incorporated into DNA.
The trinucleotide repeat disorders are divided into 2 categories determined by the type of repeat. The most common repeat is the triplet CAG which when present in the coding region of a gene codes for the amino acid glutamine (Q). Therefore, these disorders are referred to as the polyglutamine (polyQ) disorders with the best known disorder of this type being Huntington disease (Clinical Box 35-1). The remaining disorders, either do not involve the CAG triplet, or the CAG triplet is not in the coding region of the gene and are, therefore, referred to as the nonpolyglutamine disorders. Fragile X syndrome (Clinical Box 35-2) is the best known form of a nonpolyglutamine expansion disorder.
Postreplicative Modification of DNA: Methylation
One of the major postreplicative reactions that modifies the DNA is methylation. The sites of natural methylation (ie, not chemically induced) of eukaryotic DNA is always on cytosine residues that are present in CpG dinucleotides. However, it should be noted that not all CpG dinucleotides are methylated at the C residue. The cytidine is methylated at the 5 position of the pyrimidine ring generating 5-methylcytidine. In a general sense what is known about DNA methylation and transcriptional status is that when regions of a gene that can be methylated are overmethylated the associated gene is transcriptionally silent, and when the region is undermethylated the gene is transcriptionally active or can be activated.
The methylation of DNA is catalyzed by several different DNA methyltransferases (DNMT). When cells divide the DNA contains 1 strand of parental DNA and 1 strand of the newly replicated DNA (the daughter strand). If the DNA contains methylated cytidines in CpG dinucleotides the daughter strand must undergo methylation in order to maintain the parental pattern of methylation. This “maintenance” methylation is catalyzed by DNMT1, and thus this enzyme is called the maintenance methylase (Figure 35-10).

FIGURE 35-10: Process of maintenance methylation of DNA following replication. Reproduced with permission of themedicalbiochemistrypage, LLC.
Several proteins bind to methylated CpG but not to unmethylated CpG. One such protein is MeCP2 (methyl Cp binding protein 2). When MeCP2 binds to methylated CpG dinucleotides the DNA takes on a closed chromatin structure and leads to transcriptional repression. The importance of MeCP2 in regulating chromatin structure and consequently transcription is demonstrated by the fact that deficiencies in this protein result in Rett syndrome (Clinical Box 35-3).
DNA Methylation and Epigenetics
The term epigenetics is used to define the mechanism by which changes in the pattern of inherited gene expression occur in the absence of alterations or changes in the nucleotide composition of a given gene. Several different types of epigenetic events have been identified and include DNA methylation. DNA methylation is likely to be the most important epigenetic event controlling and maintaining the pattern of gene expression during development.
Whereas epigenesis plays a vital role in the regulation, control, and maintenance of gene expression leading to the many differentiation states of cells in an organism, recent evidence has identified a linkage between epigenetic processes and disease. Most significant is the link between epigenesis and cancer which has been suggested to be a contributing factor in nearly half of all cancers. A clear demonstration has been made between changes in the methylation status of tumor suppressor genes and the development of many types of cancers.
CLINICAL BOX 35-1: HUNTINGTON DISEASE
Huntington disease (HD) is an autosomal dominantly inherited disease characterized by slowly progressive neurodegeneration associated with choreic movements (abnormal involuntary movements) and dementia. The pathology of HD reveals neurodegeneration in the corpus striatum and shrinkage of the brain. HD is caused by expansion of a CAG trinucleotide repeat in the first exon of the huntingtin gene. The CAG repeat resides 17 codons downstream of the initiator AUG codon. The range of CAG repeat size in normal individuals is 6 to 34 and in affected individuals it is 36 to 121. Repeat growth from a premutation length (29–35) to the disease range (>35) occurs almost exclusively through paternal transmission. The longer the length of the CAG repeat, the earlier is the onset of symptoms. However, there is a wide degree of variation in age of onset for any given length of CAG repeat. Thus, the CAG repeat number itself is not predictive for age of onset. The variation in age of onset is related to the effects of other modifying genes, the environment, and the length of the CAG repeat. In addition, HD exhibits a genetic phenomenon termed “anticipation” which means that the symptoms of the disease appear earlier and are more severe in subsequent generations. This phenomenon is explained by meiotic instability which increases the CAG repeat number and is greater in spermatogenesis than in oogenesis. Therefore, anticipation is mainly observed when the mutation is inherited through the paternal line. Wild-type huntingtin protein is found primarily in the cytoplasm with a small percentage being intranuclear. The protein is associated with the plasma membrane, the ER, the Golgi apparatus, endocytic vesicles, the mitochondria, and microtubules. Although the exact function of Huntingtin is still poorly defined, it is known that the protein is essential for early embryonic development, is important in adult neurons and testes for cellular viability, and has a role in regulation of transcription through its interaction with numerous transcription factors and other proteins involved in the regulation of mRNA production. The classical symptoms of Huntington disease consist of progressive dementia, evolving involuntary movements and psychiatric disturbances that include personality changes and mood disorder. Choreic movements are the most prominent physical abnormality in HD and as such the disease was early on referred to as Huntington’s chorea. The earliest indications of HD are the appearance of spasmodic twitching of the extremities, generally beginning with the fingers. Additional early physical manifestations of HD are clumsiness, hyperreflexia, and eye movement disturbances. Positron-emission tomography (PET scanning) is used to demonstrate a loss of uptake of glucose in the caudate nuclei and is a valuable indication of affectation in the presymptomatic period in HD patients. There is no cure for HD and all afflicted individuals will succumb to the disease. The age of death in HD is related to the age of onset, that is, the earlier the onset, the earlier an individual will die.
CLINICAL BOX 35-2: FRAGILE X SYNDROME
Fragile X syndrome is an X-linked dominant disorder representing the most common form of inherited mental retardation. The syndrome belongs to a family of disorders that are related to the relationship between fragile chromosomal sites and disease. Fragile chromosomal sites are only detectable in vitro when cells are exposed to chemical agents that disrupt the DNA replication process. Fragile X syndrome occurs with a frequency of approximately 1 in 4000 males and 1 in 8000 females. Affected males will exhibit moderate to severe mental retardation as well as a speech delay, hyperactivity, and behavioral and social difficulties. Fragile X syndrome results from the expansion of a CGG trinucleotide repeat in the FMR1 gene which encodes the FMRP protein. The CGG repeat resides in the 5′-untranslated region (UTR) of the FMR1 gene. Normal individuals have 6 to 60 CGG repeats and the premutation size is between 55 and 200 repeats. A full mutation in affected individuals is more than 200 repeats. Repeat expansion from the premutation length to a full-mutation length occurs almost exclusively through maternal transmission. Males with the premutation–sized expansion develop a late-onset neurodegenerative condition termed fragile X-associated tremor/ataxia syndrome (FXTAS). In females with the premutation expansion there is a 25% chance of their developing ovarian failure.
The encoded FMRP protein is an RNA-binding protein found in ribonucleoprotein complexes associated with polyribosomes. Early during human fetal development the FMR1 gene is expressed at highest levels in cholinergic neurons of the nucleus basalis magnocellularis and in pyramidal neurons of the hippocampus which may contribute to the pathogenesis of the syndrome. One important pathway involving the activity of FMRP is the metabotropic glutamate receptor-(mGluR) dependent long-term depression (LTD) pathway. Glutamate functions in both the central and peripheral nervous systems are involved in memory, learning, anxiety, and the perception of pain. The characteristic symptoms of fragile X syndrome in males are moderate to severe mental retardation. In males with the full mutation (>200 repeats) the physical hallmarks of the disorder include a long narrow face, prominent ears, and macroorchidism (large testicles). These physical signs are most apparent in postpubertal males. Additional physical findings can include hyperextensible finger joints, flat feet, double jointed thumbs, and a high arched plate. Fragile X syndrome also manifests with distinct behavioral abnormalities that include hyperactivity, anxiety, extreme sensitivity to stimulation and sensations, hyperarousal, and occasionally aggressive tendencies. The behavioral anomalies overlap with those of autism such as impaired verbal and nonverbal communication, difficulty with social interactions, poor eye contact, tactile defensiveness, hand flapping, and hand biting. Females with a full mutation are less affected with respect to all aspects of the mental and physical characteristics of fragile X syndrome due to the process of X chromosome inactivation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


