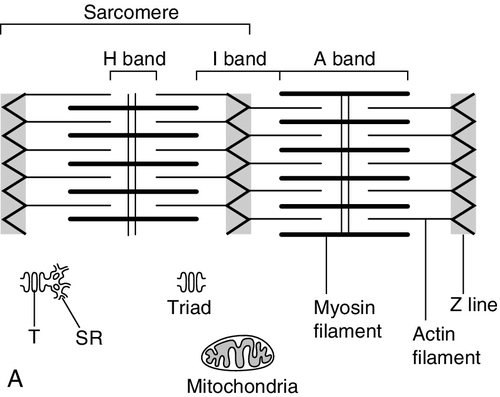
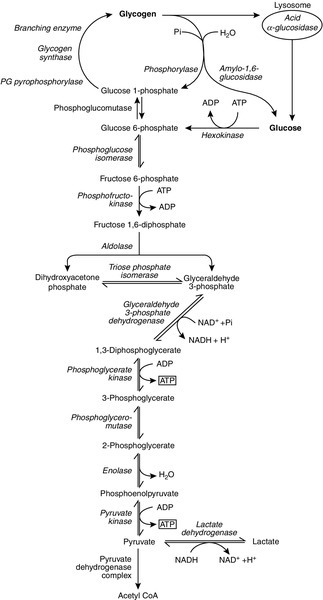
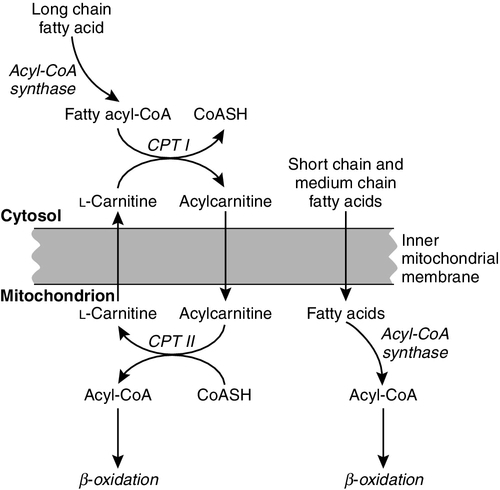
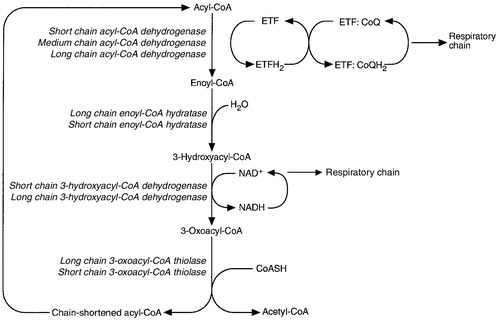
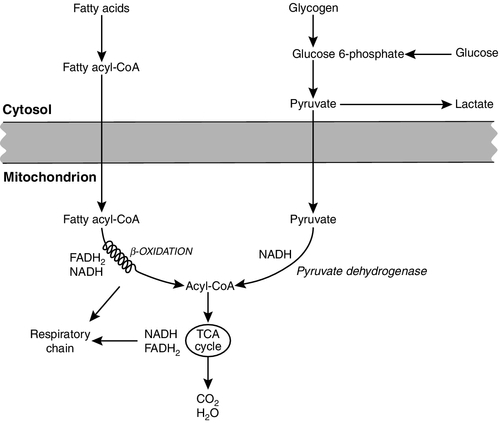
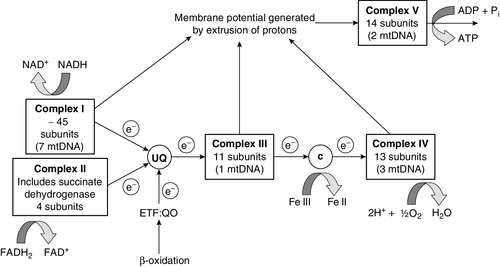
CHAPTER 33 CHAPTER OUTLINE FUNCTIONAL ANATOMY AND PHYSIOLOGY OF MUSCLE DISEASES OF MUSCLE AND THEIR INVESTIGATION BIOCHEMICAL INVESTIGATION OF MUSCLE DISEASE Plasma creatine kinase activity Other enzymes measurable in plasma INVESTIGATION OF MUSCLE DISEASE Non-metabolic, genetically determined myopathies Metabolic, genetically determined myopathies Diseases affecting striated muscle are important causes of morbidity and mortality. They are common in clinical practice, and patients may be seen by a variety of clinical specialists, including neurologists, rheumatologists, orthopaedic surgeons and paediatricians. Investigating suspected muscle disease requires a combination of clinical and laboratory skills, including biochemical, genetic and pathological investigations, each assuming a different importance depending on the nature of the disorder. In some, biochemical studies play a minor role whereas in others, particularly the metabolic myopathies, biochemical investigations are crucial. Skeletal muscle accounts for approximately 40% of total body weight and between 30% and 40% of total body oxygen consumption, even at rest. It is, therefore, an extremely important tissue in metabolic terms. Muscle is composed of multinucleated fibres that contain the contractile apparatus upon which movement depends. Although similar in structure, muscle fibres vary and three main types have been defined using metabolic and functional criteria (Fig. 33.1 and Table 33.1). Most skeletal muscles contain all three fibre types, although the proportions vary considerably depending on the function of the particular muscle. FIGURE 33.1 Cross-section of normal muscle showing the histochemical reaction for ATPase activity. The three fibre types can be easily identified: type I (dark), type IIa and type IIb. (Photograph courtesy of Dr M A Johnson.) The main function of muscle is to generate force in a controlled manner. This force, in the form of contraction (shortening), is produced in muscle fibres by the interaction of actin and myosin (Fig. 33.2), a process that is highly energy dependent. The energy required for muscle contraction comes from the hydrolysis of ATP, and maintenance of ATP concentration is critical. Any interference with ATP generation will inevitably impair the ability of muscle to produce force. ATP concentration is maintained by one of two mechanisms: ATP can be regenerated either from the energy storage molecule phosphocreatine or from ADP, or produced directly during glycolysis and mitochondrial oxidation. Regeneration is rapid, while the second process takes longer. FIGURE 33.2 The structure of skeletal muscle. (A) Diagrammatic representation of the structural components within a single muscle fibre. Thin filaments (actin) are anchored to the Z line. Thick filaments are composed of multiple myosin molecules, each of which has a hinged end that can interact with the thin filament. The T tubule is a continuation of the sarcolemmal membrane, which interacts with the sarcoplasmic reticulum at specific sites (triad). The signal for contraction is transmitted along the sarcolemma to the T tubule, which in turn causes release of calcium from the sarcoplasmic reticulum (SR). Calcium release stimulates contraction. (B) Electron micrograph of normal muscle. This shows the bundles of myofilaments with Z lines (z), thin (I band) and thick (A band) filaments. Triad (Tr) and mitochondria (m) are identified. (Photograph courtesy of Dr M J Cullen. Magnification × 30 000.) Phosphocreatine is present in large quantities in muscle (the other major site is brain) and acts as a reservoir of high-energy phosphate groups that it can donate to ADP in the following transphosphorylation reaction, catalysed by the enzyme creatine kinase: The concentration of ATP does not fall significantly until nearly all of the phosphocreatine has been converted to creatine. A second transphosphorylation reaction, catalysed by adenylate kinase, seems to have a minor role in ATP production. The AMP formed is broken down further by AMP deaminase. ATP can also be generated directly by glycolysis and the oxidative catabolism of carbohydrate and lipid fuels. These processes are slow compared with transphosphorylation, but nevertheless are essential for ATP generation. The breakdown of carbohydrate by glycolysis (Fig. 33.3) has a vital role in skeletal muscle since it permits ATP production under anaerobic conditions. When oxidative metabolism of pyruvate is impaired, for example during ischaemia, which includes high intensity activity, or in the presence of a defect of the respiratory chain, increasing amounts of lactate may be produced. FIGURE 33.3 Glycogen breakdown and glycolysis. Under anaerobic conditions, when further oxidation of acetyl-CoA is impaired, pyruvate is metabolized to lactate. This will also occur in defects of pyruvate dehydrogenase complex (PDC) and of the respiratory chain, and is due to the failure to reoxidize intramitochondrial NADH, which in turn inhibits PDC. NADH formed during glycolysis (and other substrate oxidation) is reoxidized by complex I of the mitochondrial respiratory chain (see Fig. 33.7). While small amounts of ATP can be generated by glycolysis in the cytosol, significantly greater amounts are produced by the oxidative breakdown of metabolic fuels (pyruvate, ketone bodies, fatty acids) that occurs within mitochondria. Long chain fatty acids, either from intracellular lipid stores or imported from the bloodstream, are first activated to their acyl-CoA esters before being transported into the mitochondrial matrix by the concerted action of carnitine palmitoyltransferase I, carnitine/acylcarnitine translocase and carnitine palmitoyltransferase II (Fig. 33.4). Short and medium chain fatty acids enter the mitochondria as the free acids and are activated to their acyl-CoA esters in the mitochondrial matrix. Inside the mitochondria, fatty acyl-CoA esters undergo β-oxidation, a series of four reactions that results in the production of acetyl-CoA and a chain-shortened fatty acid (Fig. 33.5); there are two or three enzymes with overlapping substrate specificities for each of these steps. Reducing equivalents generated by the process of β-oxidation are transferred to the respiratory chain. The acyl-CoA dehydrogenases transfer reducing equivalents to electron transfer flavoprotein (ETF) and thereafter to ETF dehydrogenase, which directly reduces ubiquinone of the respiratory chain (see Fig. 33.7). The 3-hydroxyacyl-CoA dehydrogenases reduce NAD+ to give NADH, which transfers its reducing equivalents to complex I of the respiratory chain. Acetyl-CoA generated either from carbohydrate or fatty acid oxidation is metabolized further by the tricarboxylic acid cycle (Fig. 33.6). The oxidation of fatty acids and glucose, as well as the subsequent metabolism of acetyl-CoA, generates more reduced cofactors (NADH and FADH2) that are re-oxidized by the respiratory chain, and the energy released by this process is conserved as ATP (Fig. 33.7). FIGURE 33.4 Transport of fatty acids across the inner mitochondrial membrane. Short and medium chain fatty acids do not require a specialized transport mechanism to cross the membrane. Long chain fatty acids are first acylated in the cytosol. Carnitine palmitoyltransferase (CPT) I on the outer side of the inner membrane converts the fatty acyl-CoA to an acylcarnitine ester and this is transported across the membrane linked to the export of carnitine. Inside the matrix, CPT II converts the acylcarnitine back to the fatty acyl-CoA, which is broken down by β-oxidation. FIGURE 33.5 Mitochondrial β-oxidation of saturated fatty acids. ETF, electron transfer flavoprotein; CoQ, coenzyme Q. FIGURE 33.6 The production and further metabolism of acetyl-CoA. FIGURE 33.7 Components of the mitochondrial respiratory chain. NADH is reoxidized by complex I, while FADH2 donates electrons via complex II and ETF dehydrogenase (ETF:QO) donates electrons directly to uniquinone (UQ). Electron transport generates sufficient energy at three sites (complexes I, III and IV) to pump protons out of the matrix, producing an electrochemical gradient. This gradient is discharged by ATP synthase (complex V) and the energy released used to drive the phosphorylation of ADP to ATP. c, Cytochrome c; e−, electron. The balance of muscle metabolism depends on the state of activity, diet and the influence of various hormones (particularly insulin, thyroxine, glucocorticoids). At rest, muscle predominantly oxidizes fatty acids to generate the energy for ATP synthesis. During exercise, the proportion of energy derived from carbohydrate or lipid depends on the degree and duration of this exercise and on the degree of physical fitness. High-intensity exercise at close to maximum oxygen uptake relies almost exclusively on carbohydrate metabolism, and glycogen depletion coincides with exhaustion. During moderate-intensity exercise for prolonged periods, there is a switch from carbohydrate to lipid metabolism. There are a large number of different disorders of muscle, and while our classification includes the main categories (Box 33.1), more comprehensive lists are available (see Karpati et al. in Further reading, below). A detailed description of the clinical features associated with the different types of muscle disease is outside the scope of this chapter, but is discussed in several texts on muscle disease. The clinical features depend upon the age of the patient and the type of disease. For instance, a child with Duchenne muscular dystrophy will experience difficulty rising from sitting or lying and may have frequent falls. Such problems will prompt the parents to seek advice. In adults, the main forms of presentation are weakness, fatigue and pain. Less commonly, muscle wasting, swelling or twitching of the muscle or a skin rash may be a first symptom. In the genetically determined disorders, the weakness is usually gradually progressive and often follows a characteristic pattern. In other myopathies, there may be associated stigmata, for instance joint disease or skin rash suggesting a connective tissue disorder; anxiety, sweating and weight loss suggesting hyperthyroidism, or features compatible with high alcohol intake. The muscle pain described by patients with muscle disease may be important in suggesting whether there may be a metabolic cause. For instance, both defects of carbohydrate metabolism and fatty acid oxidation will cause muscle pain associated with exercise. The pain associated with defects of carbohydrate metabolism occurs during high intensity exercise when glycolysis generates most of the energy required for muscle contraction, whereas defects of fatty acid oxidation cause muscle pain after prolonged exercise at a time when fatty acids are the predominant metabolic fuels. The clinician must evaluate the clinical features and decide which investigations are appropriate. In many patients with suspected muscle disease, this will involve a combination of biochemical, molecular genetic, neurophysiological and morphological investigations. While many biochemical and genetic studies are performed on blood samples, morphological study and biochemical analyses such as measurement of muscle enzyme activity, require tissue. Muscle biopsy is a relatively simple procedure and there are two main methods: an open biopsy, in which relatively large amounts (0.5–3 g) of muscle can be removed, and a needle biopsy, in which smaller amounts (50–200 mg) are obtained. For most biochemical and histochemical studies, small amounts are sufficient. Morphological changes alone may be sufficient to suggest a diagnosis, for example of Duchenne muscular dystrophy. The diagnosis of metabolic myopathies has been greatly improved by the development of cytochemical techniques that can show, for example, the abnormal storage of glycogen or lipid, or demonstrate the presence or absence of specific enzyme activities in situ. A further development in this area is the use of specific antisera to enable the precise localization (and therefore the presence or absence) of proteins at the cellular level. This technique of immunocytochemistry provides valuable additional information in the investigation of muscle disease. These include the measurement of plasma sodium, potassium, chloride, urea, bicarbonate, glucose, calcium and phosphate, together with simple tests of endocrine function. While not all these tests are necessary for each patient with muscle disease, disturbances of each parameter can result in muscle disease, as shown by the following examples. Severe hypokalaemia associated, for example, with diuretic use or liquorice ingestion, can result in muscle weakness. Renal failure may lead to muscle weakness for several reasons, including electrolyte disturbance and altered calcium metabolism. It must also be remembered that acute muscle necrosis from any cause (e.g. malignant hyperpyrexia, drugs, injury or metabolic myopathy) may itself cause acute kidney injury owing to the tubulotoxic effect of myoglobin. Muscle symptoms are common in endocrine disturbances: hypothyroidism, for example, may be associated with proximal weakness, often with discomfort in the affected muscles. The measurement of plasma enzyme activity is important in the diagnosis of muscle disease, and while the activities of several enzymes may be elevated, creatine kinase (CK) is the most sensitive indicator of muscle damage. Skeletal muscle has the highest CK content of any tissue, more than three times as much as heart or brain, and consequently nearly all CK activity in normal plasma is derived from skeletal muscle. In addition, CK activity is more frequently abnormal than other enzymes in neuromuscular disease and the range of abnormal values is greater.
Muscle disease
INTRODUCTION
FUNCTIONAL ANATOMY AND PHYSIOLOGY OF MUSCLE





DISEASES OF MUSCLE AND THEIR INVESTIGATION
BIOCHEMICAL INVESTIGATION OF MUSCLE DISEASE
‘Routine’ biochemical studies
Plasma creatine kinase activity

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


