CHAPTER OUTLINE
Metabolic Classifications of Amino Acids
Glutamate and Glutamine Metabolism
Aspartate and Asparagine Metabolism
Alanine and the Glucose-Alanine Cycle
Cysteine and Methionine Metabolism
Arginine, Ornithine, and Proline Metabolism
Phenylalanine and Tyrosine Metabolism
Leucine, Isoleucine, and Valine Catabolism: Branched-Chain Amino Acids
High-Yield Terms
Glucogenic: relating to amino acids whose catabolic products can be used for net glucose synthesis during gluconeogenesis
Ketogenic: relating to amino acids whose catabolic products enter the tricarboxylic acid (TCA) cycle but cannot be used for net glucose synthesis during gluconeogenesis
Essential amino acid: any amino acid that cannot be formed de novo or from other precursor compounds within mammalian cells
Glutamate dehydrogenase: critical enzyme catalyzing the interconversion of glutamate and α-ketoglutarate, serves as a gateway reaction between cellular energy demands and nitrogen homeostasis
Asparaginase: enzyme catalyzing the deamination of asparagine to aspartate, enzyme is used clinically as a chemotherapeutic treatment for certain leukemias
Cystathionine β-synthase: enzyme involved in cysteine biosynthesis, deficiencies are most common cause of homocystinurias
Glycine cleavage complex: also called glycine decarboxylase, enzyme-catabolizing glycine, defects in activity cause glycine encephalopathy
Phenylketonuria (PKU): a particular form of hyperphenylalaninemia resulting from defects in the phenylalanine hydroxylase gene
Branched-chain amino acid: consists of leucine, isoleucine, and lysine; defects in the catabolic enzyme branched-chain keto acid dehydrogenase (BCKD) result in Maple syrup urine disease (MSUD)
Alkaptonuria: first inherited error in metabolism to be characterized, benign disease caused by defects in the tyrosine-catabolizing enzyme homogentisate oxidase, urine of afflicted patients turns brown on exposure to air
Amino Acid Uptake
All tissues have some capability for synthesis of the nonessential amino acids, amino acid remodeling, and conversion of nonamino acid carbon skeletons into amino acids and other derivatives that contain nitrogen. However, many amino acids cannot be synthesized by mammalian cells nor derived from other precursors and so must be acquired from the diet. The process of protein digestion followed by peptide and amino acid uptake by the gut is described in detail in Chapter 43. Briefly, proteins are degraded to free amino acids, di- and tripeptides by gut and pancreatic peptidases. Intestinal uptake of amino acids involves several different Na+-dependent amino acid transporters present in the apical membrane of intestinal luminal brush border cells. These include acidic, basic, and neutral amino acid transporters, as well as a proline and glycine transporter. The inability to effectively absorb amino acids from the gut can lead to serious developmental and physiologic consequences, such as in the case of Hartnup disorder (Clinical Box 30-1).
Metabolic Classifications of Amino Acids
In times of both dietary surplus and fasting/starvation, the potentially toxic nitrogen of amino acids is eliminated via transaminations, deamination, and urea formation; the carbon skeletons are generally conserved as carbohydrate, via gluconeogenesis, or as fatty acid via fatty acid synthesis pathways. In this respect, amino acids fall into 3 categories: glucogenic, ketogenic, or glucogenic and ketogenic. Glucogenic amino acids are those that give rise to a net production of pyruvate or TCA cycle intermediates, such as α-ketoglutarate or oxaloacetate, all of which are precursors to glucose via gluconeogenesis. All amino acids except lysine and leucine are at least partly glucogenic. Lysine and leucine are the only amino acids that are solely ketogenic, giving rise only to acetyl-CoA or acetoacetyl-CoA, neither of which can bring about net glucose production.
A small group of amino acids comprised of isoleucine, phenylalanine, threonine, tryptophan, and tyrosine give rise to both glucose and fatty acid precursors and are thus characterized as being glucogenic and ketogenic. Finally, it should be recognized that amino acids have a third possible fate. During times of starvation, the reduced carbon skeleton is used for energy production, with the result that it is oxidized to CO2 and H2O (Table 30-1).

CLINICAL BOX 30-1: HARTNUP DISORDER
Hartnup disorder was first described in 1956 in the Hartnup family in London as a renal aminoaciduria of neutral amino acids associated with a pellagra-like skin rash and episodes of cerebellar ataxia. The disorder is caused by a defect in neutral amino acid transport in the apical brush border membranes of the small intestine and in kidney proximal tubules. The transporter is a member of the solute carrier family, specifically the SLC6A19 transporter. SLC6A19 is also known as the system B(0)–neutral amino acid transporter 1 (B[0]AT1). SLC6A19 is responsible for the transport of neutral amino acids in a Na+-dependent transport reaction. The lack of intestinal tryptophan transport is responsible for most if not all clinical phenotypes of Hartnup disorder. The pellagra-like skin rash seen on sun-exposed areas of skin in Hartnup disorder patients is most likely the result of nicotinamide deficiency due to a lack of tryptophan, which is a precursor for its synthesis. Symptoms of Hartnup disorder may begin in infancy or early childhood, but sometimes they begin as late as early adulthood. Symptoms may be triggered by sunlight, fever, drugs, or emotional or physical stress. Most symptoms occur sporadically and are caused by a deficiency of niacin. When Hartnup disorder manifests during infancy, the symptoms can be variable in clinical presentation. These symptoms include failure to thrive, photosensitivity, intermittent ataxia, nystagmus, and tremor.
The amino acids arginine, methionine, and phenylalanine are considered essential for reasons not directly related to lack of synthesis. Arginine is synthesized by mammalian cells but at a rate that is insufficient to meet the growth needs of the body and the majority that is synthesized is cleaved to form urea. Methionine is required in large amounts to produce cysteine if the latter amino acid is not adequately supplied in the diet. Similarly, phenylalanine is needed in large amounts to form tyrosine if the latter is not adequately supplied in the diet.
Glutamate and Glutamine Metabolism
Glutamate is synthesized by the reductive amination of α-ketoglutarate catalyzed by glutamate dehydrogenase (see Figure 29-3). As discussed in the Chapter 29, the glutamate dehydrogenase reaction plays a central role in overall nitrogen homeostasis. In addition, glutamate is synthesized from α-ketoglutarate via the action of several different aminotransferases, with the amino nitrogen being donated by a number of different amino acids. In this capacity, glutamate serves as the major “transporter” of waste nitrogen during amino acid catabolism.
Glutamine is produced from glutamate by a transamidation reaction catalyzed by the ATP-dependent enzyme glutamine synthetase (see Figure 29-4).
Aminotransferase reactions are readily reversible. The direction of any individual transamination depends principally on the concentration of reactants and products. By contrast, transamidation reactions, which are dependent on ATP, are considered irreversible. As a consequence, the degradation of glutamine takes place by a hydrolytic pathway rather than by a reversal of the pathway by which it was formed.
Glutaminase (see Figure 29-5) is an important kidney tubule enzyme involved in converting glutamine (primarily from liver) to glutamate and NH4+, with the NH4+ being excreted in the urine. Glutaminase activity is present in many other tissues as well, although its activity is not nearly as prominent as in the kidney. Of particular significance to renal control of blood pH, the glutamate produced from glutamine can be catabolized to α-ketoglutarate and another mole of NH4+ via the action of glutamate dehydrogenase. The α-ketoglutarate can serve as precursor for renal gluconeogenesis (see Chapter 13), making glutamine a glucogenic amino acid.
High-Yield Concept
Glutamine represents the major nitrogen-carrying compound in the blood and as such plays a critical role in overall nitrogen homeostasis (see Chapter 29).
Aspartate and Asparagine Metabolism
Like glutamate, aspartate is synthesized by a simple 1-step transamination reaction catalyzed by aspartate aminotransferase (AST) (Figure 30-1). This reaction uses the aspartate α-keto acid analog, oxaloacetate (OAA), and glutamate as the amino donor. AST is also historically referred to as serum glutamate-oxalate transaminase (SGOT). The transfer of nitrogen to OAA is an important component of substrate generation (aspartate) for the production of ornithine in the urea cycle (see Chapter 29).

FIGURE 30-1: Aspartate aminotransferase (AST) reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.
Asparagine is synthesized from aspartate via the action of asparagine synthetase (Figure 30-2) which, like glutamine synthetase, is an ATP-dependent aminotransferase reaction. Aspartate can also be formed by deamination of asparagine catalyzed by asparaginase (Figure 30-3). Asparaginase has utility as an anti-cancer drug due to the role of asparagine in hematopoietic cancers (Clinical Box 30-2).

FIGURE 30-2: Asparagine synthetase reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.

FIGURE 30-3: Asparaginase reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.
Alanine and the Glucose-Alanine Cycle
Aside from its role in protein synthesis, alanine is second only to glutamine in prominence as a circulating amino acid. In this capacity, it serves a unique role in the transfer of nitrogen from peripheral tissues to the liver. Alanine is transferred to the circulation by many tissues, but mainly by muscle, in which alanine is formed from pyruvate at a rate proportional to intracellular pyruvate levels. Liver accumulates plasma alanine, reverses the transamination that occurs in muscle, and proportionately increases urea production. The pyruvate is either oxidized (see Chapter 16) or converted to glucose via gluconeogenesis (see Chapter 13). When alanine transfer from muscle to liver is coupled with glucose transport from liver back to muscle, the process is known as the glucose-alanine cycle (see Figure 13-2).
There are 2 main pathways to production of alanine: directly from protein degradation, and via the transamination of pyruvate by alanine transaminase (ALT) (Figure 30-4). ALT is also historically referred to as serum glutamate-pyruvate transaminase (SGPT).

FIGURE 30-4: Alanine transaminase (ALT) reaction. Reproduced with permission of themedicalbiochemistrypage, LLC.
Cysteine and Methionine Metabolism
Cysteine and methionine metabolism are interrelated as the sulfur for cysteine synthesis comes from methionine (Figure 30-5). In cysteine synthesis, homocysteine condenses with serine to produce cystathionine, which is subsequently cleaved by cystathionase to produce cysteine and α-ketobutyrate. The sum of the latter 2 reactions is known as trans-sulfuration. The α-ketobutyrate from this reaction is first converted to propionyl-CoA and then via a 3-step process (see Figure 25-4) to the TCA cycle intermediate, succinyl-CoA. Other than protein, the most important product of cysteine metabolism is the bile salt precursor taurine, which is used to form the bile acid conjugates, taurocholate and taurochenodeoxycholate (see Chapter 27).
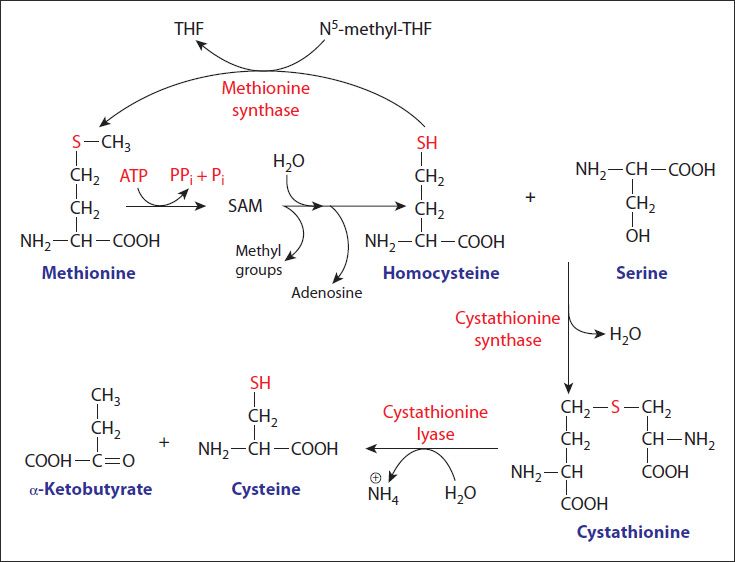
FIGURE 30-5: Utilization of methionine in the synthesis of cysteine. Reproduced with permission of themedicalbiochemistrypage, LLC.
CLINICAL BOX 30-2: THERAPEUTIC USE OF ASPARAGINASE
Asparaginase is used as a chemotherapeutic drug in the treatment of acute lymphocytic leukemias (ALL). The rationale for the use of asparaginase in this capacity stems from the fact that ALL cells are unable to synthesize asparagine, whereas normal cells are able to make their own asparagine. Therefore, leukemic cells require high amounts of circulating asparagine for protein synthesis and growth. Since asparaginase catalyzes the conversion of asparagine to aspartic acid and ammonia, the leukemic cells are deprived of circulating asparagine. The inability to acquire sufficient asparagine leads to the activation of amino acid deprivation responses in the leukemic cells, which culminates with the triggering of programmed cell death pathways in these cells (see Chapter 52).
The 2 key enzymes of this pathway, cystathionine β-synthase (CBS) and cystathionase (cystathionine lyase), use pyridoxal phosphate as a cofactor. In addition, both enzymes are under regulatory control. Cystathionase is negatively regulated by the allosteric effector, cysteine. Cysteine also inhibits the expression of the CBS gene.
Genetic defects have been identified in both the CBS and the cystathionase genes. Missing or impaired CBS leads to homocystinuria (Clinical Box 30-3). Missing or impaired cystathionase leads to excretion of cystathionine in the urine but does not have any other untoward effects. Rare cases have been found in which cystathionase is defective and operates at a low level, resulting in methioninuria with no other consequences.
CLINICAL BOX 30-3: HOMOCYSTINURIA
Homocystinurias represent a family of inherited disorders resulting from defects in several of the genes involved in the conversion of methionine to cysteine. As the name implies, these disorders result in elevated levels of homocysteine in the urine. In addition, patients excrete elevated levels of methionine and metabolites of homocysteine. The most common causes of homocystinuria are defects in the cystathionine β-synthase (CBS) gene. Homocystinuria is often associated with mental retardation, although the complete syndrome is multifaceted and many individuals with this disease are mentally normal, while others experience variable levels of developmental delay along with learning problems. Common symptoms of homocystinuria are dislocated optic lenses, osteoporosis, lengthening and thinning of the long bones, and an increased risk of abnormal blood clotting (thromboembolism). Some instances of genetic homocystinuria respond favorably to pyridoxine therapy, suggesting that in these cases the defect in CBS is a decreased affinity for the cofactor, pyridoxal phosphate.
Homocystinuria can also result from vitamin deficiencies due to the role of the co-factor forms of B6, B12, and folate in the overall metabolism of methionine. Vitamin B6 (as pyridoxal phosphate) is required for the activity of CBS and the homocystinuria that results with B6 deficiency is also associated with elevated methionine levels in the blood. Vitamin B12 and folate (as N5-methyl-THF) are required for the methionine synthase reaction so a deficiency of either vitamin can result in homocystinuria presenting with reduced levels of plasma methionine. The enzyme methylmalonyl-CoA mutase (see Chapter 25) also requires B12 and so a homocystinuria resulting from a deficiency in this vitamin is also associated with methylmalonic academia. Indeed, the measurement of serum methionine and methylmalonic acid in cases of homocystinuria allows for a differential diagnosis of the nutritional (non-genetic) cause.
Cystathionase can also transfer sulfur from one cysteine to another, generating thiocysteine and pyruvate. Transamination of cysteine yields β-mercaptopyruvate, which then reacts with sulfite (SO32−), to produce thiosulfate (S2O32−) and pyruvate. Both thiocysteine and thiosulfate can be used by the enzyme rhodanese to incorporate sulfur into cyanide (CN−), thereby detoxifying the cyanide to thiocyanate.
While cysteine readily oxidizes to form the disulfide cystine, cells contain little if any free cystine. The cystine is rapidly converted to cysteine in a reaction catalyzed by cystine reductase (Figure 30-6). An additionally important enzyme activity utilizes glutathione in the reduction of cystine to cysteine. In this reaction, catalyzed by glutathione-cystine transhydrogenase, glutathione (GSH) is oxidized to glutathione disulfide (GSSG) while cystine is reduced to cysteine.

FIGURE 30-6: Reduction of cystine to cysteine in the cystine reductase reaction. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
High-Yield Concept
Elevated levels of homocysteine in the blood have been shown to correlate with cardiovascular dysfunction. The role of homocysteine in cardiovascular disease is related to its ability to induce a state of inflammation. Homocysteine serves as a negatively charged surface that attracts the contact phase of the intrinsic pathway of blood coagulation (see Chapter 51). Activation of the intrinsic coagulation cascade leads to inappropriate thrombolytic events as well as resulting in increases in inflammatory cytokine release from leukocytes that are activated as a result of the procoagulant state.
There are several pathways for cysteine catabolism. The major catabolic pathway in animals is via cysteine dioxygenase that oxidizes the cysteine sulfhydryl to sulfonate, producing the intermediate cysteine sulfonate (Figure 30-7). Cysteine sulfonate can serve as a biosynthetic intermediate undergoing decarboxylation and oxidation to produce taurine used in bile acid metabolism. Catabolism of cysteine sulfonate yields bisulfite (HSO3−) and pyruvate. Sulfite oxidase converts HSO3− to sulfate, SO4− and H2O2 with the sulfate serving as a substrate for the formation of 3′-phosphoadenosine-5′-phosphosulfate (PAPS). PAPS is used for the transfer of sulfate to biological molecules such as the sugars of the glycosphingolipids (see Chapter 21).

FIGURE 30-7: Two pathways catabolize L-cysteine: the cysteine sulfinate pathway (top) and the 3-mercaptopyruvate pathway (bottom). Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
In additional to its role as a precursor for cysteine synthesis and as an amino acid for protein synthesis, methionine serves an important role in numerous methylation reactions in the form of S-adenosylmethionine, SAM (also abbreviated AdoMet). The synthesis of SAM is catalyzed by methionine adenosyltransferase (Figure 30-8). The most important reactions involving SAM as methyl donor are those of catecholamine synthesis (see Chapter 31). The result of methyl transfer is the conversion of SAM to S-adenosylhomocysteine. S-adenosylhomocysteine is then cleaved by adenosylhomocysteinase to yield homocysteine and adenosine. Homocysteine can be converted back to methionine by methionine synthase (see Figure 30-5). The principal catabolic fate of methionine is the conversion to cysteine.

FIGURE 30-8: Formation of S-adenosylmethionine. ~CH3 represents the high group transfer potential of “active methionine.” Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
Glycine and Serine Metabolism
Glycine is involved in many anabolic reactions other than protein synthesis, including the synthesis of purine nucleotides, heme, glutathione, creatine, and serine. The main pathway to glycine synthesis is a 1-step reversible reaction catalyzed by serine hydroxymethyltransferase (SHMT). This enzyme is a member of the family of 1-carbon transferases and is also known as glycine hydroxymethyltransferase (Figure 30-9). This reaction involves the transfer of the hydroxymethyl group from serine to tetrahydrofolate (THF), producing glycine and N5,N10-methylene-THF. There are mitochondrial and cytosolic versions of serine hydroxymethyltransferase. The cytosolic enzyme is referred to as SHMT1 and the mitochondrial enzyme is SHMT2.

FIGURE 30-9: Interconversion of serine and glycine by serine hydroxymethyltransferase. (H4 folate, tetrahydrofolate). Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
The main pathway to de novo biosynthesis of serine utilizes the glycolytic intermediate 3-phosphoglycerate (Figure 30-10). An NADH-linked dehydrogenase converts 3-phosphoglycerate into a keto acid, 3-phosphopyruvate, suitable for subsequent transamination. Aminotransferase activity with glutamate as a donor produces 3-phosphoserine, which is converted to serine by phosphoserine phosphatase. Serine can also be derived from glycine (and vice versa) via the SHMT-catalyzed reaction (Figure 30-11).

FIGURE 30-10: Pathway of de novo serine biosynthesis. Reproduced with permission of themedicalbiochemistrypage, LLC.

FIGURE 30-11: Reaction catalyzed by glycine decarboxylase. Reproduced with permission of themedicalbiochemistrypage, LLC.
The catabolism of serine occurs via its conversion first to glycine. Glycine can then be oxidized by glycine decarboxylase (also referred to as the glycine cleavage complex, GCC) to yield ammonia, CO2, and a second equivalent of N5,N10-methylene-THF. Glycine decarboxylase is a mitochondrial enzyme complex composed of 4 distinct proteins identified as H, L, P, and T. The H protein is a lipoic acid–containing protein. The L protein is a dihydrolipoamide dehydrogenase (DLD) similar to the DLD subunit of the pyruvate dehydrogenase and α-ketoglutarate (2-oxoglutarate) dehydrogenase complexes. The P protein is a pyridoxal phosphate-dependent glycine decarboxylase. The T protein is a tetrahydrofolate-requiring aminotransferase. Loss of activity of glycine decarboxylase (GCC) is associated with severe mental retardation (Clinical Box 30-4).
CLINICAL BOX 30-4: GLYCINE DECARBOXYLASE DEFICIENCY
Nonketotic hyperglycinemia (NKH) is an autosomal recessive inborn error of glycine metabolism due to deficiencies in the H, P, or T proteins of the glycine decarboxylase complex. Mutations in the P protein are the most common. NKH, also known as glycine encephalopathy, is characterized by severe mental retardation that is due to highly elevated levels of glycine in the CNS. Infants generally present with severe lethargy, seizures, and respiratory depression requiring mechanical ventilation. A diagnosis of NKH associated with these symptoms is made secondary to elevated plasma and cerebrospinal fluid glycine concentrations. The prognosis for infants with NKH is poor, with severe neurologic impairment, intractable seizures, and death common before 5 years of age.
Glycine is coreleased with γ-aminobutyric acid (GABA) (see Chapter 31), which is the primary inhibitory neurotransmitter. Glycine action as a neurotransmitter is a function of the amino acid binding to a specific receptor, GlyR. GlyR is a member of the nicotinic acid receptor superfamily that includes the GABAA receptor (GABAAR), the excitatory nicotinic acetylcholine receptors (nAChR), and the serotonin type 3 receptor (5HT3). GlyR is a ligand-gated inotropic receptor that is a chloride channel. In addition to glycine, GlyR can be activated by several other small amino acids such as alanine and taurine. Glycine is also involved in the modulation of excitatory neurotransmission exerted via glutamate binding to N-methyl-d-aspartate (NMDA) receptors.
In addition to its role in proteins synthesis and the formation of serine, glycine functions in the central nervous system as a major inhibitory neurotransmitter. In this capacity, glycine participates in regulating signals that process motor and sensory information that permits movement, vision, and audition.
Arginine, Ornithine, and Proline Metabolism
The synthesis and catabolism of arginine in mammals takes place within the context of the urea cycle (see Chapter 29). Synthesis occurs from citrulline and aspartate via the actions of argininosuccinate synthetase and argininosuccinate lyase. If arginine is needed for protein synthesis, it can be diverted away from the urea cycle, otherwise it is subsequently catabolized to urea and ornithine via the action of arginase.
Glutamate is the precursor of both proline and ornithine (Figure 30-12), with glutamate semialdehyde being a branch point intermediate leading to one or the other of these 2 products. While ornithine is not one of the 20 amino acids used in protein synthesis, it plays a significant role as the acceptor of carbamoyl phosphate in the urea cycle. Ornithine serves an additional important role as the precursor for the synthesis of the polyamines. The production of ornithine from glutamate is important when dietary arginine, the other principal source of ornithine, is limited.

FIGURE 30-12: Synthesis of ornithine and proline. Reproduced with permission of themedicalbiochemistrypage, LLC.
The fate of glutamate semialdehyde depends on prevailing cellular conditions. When arginine concentrations become elevated, the ornithine contributed from the urea cycle plus that from glutamate semialdehyde inhibit the aminotransferase reaction, with accumulation of the semialdehyde as a result. The semialdehyde spontaneously cyclizes to Δ1-pyrroline-5-carboxylate, which is then reduced to proline by an NADPH-dependent reductase. Likewise, ornithine, in excess of urea cycle needs, is diverted to proline synthesis or it can be converted to glutamate. The glutamate can then be converted to α-ketoglutarate in a transamination reaction. Thus arginine, ornithine, and proline are glucogenic.
Phenylalanine and Tyrosine Metabolism
Phenylalanine normally has only 2 fates: incorporation into protein and production of tyrosine via the tetrahydrobiopterin-requiring phenylalanine hydroxylase (PAH) (Figure 30-13). Thus, phenylalanine catabolism always follows the pathway of tyrosine catabolism. Tyrosine serves several important functions, including as an amino acid for protein synthesis and as an intermediate in the biosynthesis of catecholamine neurotransmitters (see Chapter 31).

FIGURE 30-13: Biosynthesis of tyrosine from phenylalanine. Reproduced with permission of themedicalbiochemistrypage, LLC.
PAH is a mixed-function oxygenase: one atom of oxygen is incorporated into water and the other into the hydroxyl of tyrosine. The tetrahydrofolate-related cofactor tetrahydrobiopterin (BH4) is required by the PAH reaction. The oxidized dihydrobiopterin (BH2) generated during the PAH reaction is reduced to BH4 by the NADH-dependent enzyme dihydropteridine reductase (DHPR).
High-Yield Concept
Missing or deficient PAH results in hyperphenylalaninemia. Hyperphenylalaninemia is defined as a plasma phenylalanine concentration greater than 2 mg/dL (120 μM). The most widely recognized hyperphenylalaninemia (and most severe) is the genetic disease known as phenylketonuria (PKU) (Clinical Box 30-5). Patients suffering from PKU have plasma phenylalanine levels greater than 1000 μM, whereas the non-PKU hyperphenylalaninemias exhibit levels of plasma phenylalanine less than 1000 μM.
CLINICAL BOX 30-5: HYPERPHENYALANINEMIAS: PKU
Phenylketonuria (PKU) is an autosomal recessive disorder resulting from defects in the metabolism of phenylalanine. PKU is a hyperphenylalaninemia with multifactorial causes. There are several hyperphenylalaninemias that are not PKU and are called non-PKU hyperphenylalaninemias (HPA). Hyperphenylalaninemia is defined as a plasma phenylalanine concentration greater than 120 μM. PKU is characterized by plasma phenylalanine greater than 1000 μM and HPA have plasma phenylalanine amounts that are less than 1000 μM. PKU is caused by mutations in the phenylalanine hydroxylase (PAH) gene. Because the reaction catalyzed by PAH involves tetrahydrobiopterin (BH4) as a cofactor, the HPA can result from defects in any of the several genes required for synthesis and recycling of BH4. Removal of excess phenylalanine normally proceeds via the tyrosine biosynthesis reaction and then via tyrosine catabolism. Because BH4 is involved in additional hydroxylation reactions, notably tryptophan and tyrosine in the brain in the formation of serotonin and the catecholamines, respectively, it is important to correctly diagnose any HPA as being the result of abnormal BH4 homeostasis or defects in PAH. The major clinical manifestation associated with PKU and many HPA is impaired cognitive development and function. The mental retardation is caused by the accumulation of phenylalanine, which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of α-ketoglutarate. This absence of α-ketoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy, which is essential to normal brain development. The product of phenylalanine transamination, phenylpyruvic acid, is reduced to phenylacetate (Figure 30-14) and all 3 compounds appear in the urine.

FIGURE 30-14: Alternative pathways of phenylalanine catabolism in phenylketonuria. The reactions also occur in normal liver tissue but are of minor significance. Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. Harper’s Illustrated Biochemistry, 29th ed. New York, NY: McGraw-Hill; 2012.
The presence of phenylacetate in the urine imparts a “mousy” odor. Currently in this country (and many others as well), newborns are routinely screened for PKU by measurement of serum phenylalanine levels. In order to classify the phenotype of PKU into severe and less severe forms, as well as to exclude BH4-deficient HPA, it is necessary to measure the plasma, urine, and cerebrospinal fluid levels of phenylalanine, pterins, and the derivatives of the neurotransmitters derived from tryptophan and tyrosine. The mainstay in treatment of PKU is the low-phenylalanine diet. Optimal treatment requires both early onset (hence the utility of the postnatal assessments in this country) and continuous treatment throughout the adolescence and possibly for life. Because of the necessity for phenylalanine in protein synthesis and neurotransmitter synthesis, it is important to carefully control the intake of the amino acid.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


