CHAPTER 15 David B. Wile; John P.H. Wilding CHAPTER OUTLINE PHYSIOLOGY AND PATHOPHYSIOLOGY OF GLUCOSE HOMOEOSTASIS Introduction: the maintenance of normoglycaemia Insulin-like growth factors and their receptors CLASSIFICATION AND DIAGNOSIS OF DIABETES MELLITUS Maturity onset diabetes of the young (MODY) ENDOCRINE ASSOCIATIONS WITH DIABETES DIABETES, NUTRITION AND GROWTH MECHANISMS OF DIABETIC TISSUE DAMAGE Other aspects of diabetic tissue damage CONDITIONS ASSOCIATED WITH INADEQUATELY CONTROLLED DIABETES MELLITUS BIOCHEMICAL MEASUREMENTS IN DIABETES MELLITUS Tests of recent glycaemic control Blood glucose concentrations are maintained within very close limits in healthy people. Any given individual has a very strictly maintained postabsorptive (e.g. fasted overnight) blood glucose concentration of 4.5–5.2 mmol/L, with intraindividual coefficients of variation (CV) of only 1–2%. Interindividual CV (assuming similar times since previous meal, levels of activity, composition of previous meal etc.) is less than 5%, so that a fasting glucose of 6.0 mmol/L is usually 4–5 standard deviations away from the mean in most populations. Glucose concentrations in healthy people increase after meals, but typical meals will not raise blood glucose above 8–10 mmol/L and normoglycaemia is usually restored within 2–4 h. Falls in glucose concentration may be produced by severe, sudden unaccustomed exercise or prolonged fasting (or both), by the pathological conditions discussed in Chapter 17 and by pharmacological means, but are not typically encountered in healthy adults on a daily basis. The strictness of glucoregulation is remarkable when compared with the relative laxity of regulation of the concentrations of other circulating metabolic fuels such as ketone bodies and non-esterified fatty acids (NEFA, also known as free fatty acids, FFA). The reason for strict avoidance of low blood glucose concentrations is readily apparent in terms of the avoidance of the neurological and other consequences of hypoglycaemia, and it may be no accident that of all the hormones known to influence blood glucose concentration, insulin is the only one able to lower it. The threshold for the onset of detectable neuroglycopaenia is of the order of 3.0–3.5 mmol/L, and it is thus appropriate that counter-regulatory mechanisms are set to respond to maintain glycaemia comfortably above this level. The reason for the strict avoidance of hyperglycaemia is less immediately apparent. Symptoms of hyperglycaemia are florid (in subjects used to relative normoglycaemia) at blood glucose concentrations of > 12–13 mmol/L and may become apparent at concentrations < 10 mmol/L. Such concentrations may be seen, for example following a myocardial infarction, so-called ‘stress hyperglycaemia’ owing to the effect of catecholamine release, and are usually transient. Stress hyperglycaemia also occurs when plasma cytokine concentrations rise, for example in sepsis, with an increase in oxidative metabolism. (The severe metabolic consequences of hyperglycaemia at concentrations usually > 20 mmol/L are discussed in the section on diabetic emergencies in Chapter 16.) In contrast, mild hyperglycaemia (glucose 6–9 mmol/L) is usually asymptomatic. The only obvious teleological value of preventing mild hyperglycaemia is the avoidance of the consequences of chronic hyperglycaemia, usually referred to as ‘long-term diabetic complications’ or ‘diabetic tissue damage’ (p. 296 and 322), and the increased susceptibility to infection that may occur acutely. The mechanisms for regulation of normoglycaemia are summarized in Box 15.1, which lists the counter-regulatory mechanisms responsible for preventing hypoglycaemia, and Figure 15.1, which outlines the main sites of insulin action relevant to prevention of hyperglycaemia. FIGURE 15.1 The principal antihyperglycaemic actions of insulin. Insulin Circulating glucose derives from three main sources: • the gut, as the result of hydrolysis or hepatic conversion of a variety of ingested carbohydrates • hepatic and some other glycogen stores (glycogenolysis) • new synthesis from precursors (gluconeogenesis). Gluconeogenesis takes place in the liver (~75–90%) and kidneys (~10–25%) from glucogenic amino acids, especially alanine, and from glycerol, lactate and pyruvate. Principally it involves the synthesis of a 6-carbon compound from various 3-carbon skeletons. Partial oxidative metabolism in tissues like adipocytes and muscle provides lactate and pyruvate but they can also be donated by red blood cells, in which anaerobic metabolism occurs in the absence of the enzymes of the Krebs (tricarboxylic acid) cycle. Triacylglycerol (triglyceride) released from adipocytes during lipolysis provides glycerol. Regulation of gluconeogenesis can be via changes in the amount of substrate arriving at the liver or by the amount that the liver itself extracts, as well as by regulation within the liver. This process is subject to direct hormonal modulation by insulin, glucagon and catecholamines and indirectly by other hormones. Hepatic glucose output is ~2.0 mg/kg body weight/min in the resting postabsorptive state, or 200–300 g during the average 24 h period (varying with the availability of glucose from food and with the body’s requirements during exercise). Plasma glucose concentration is determined by the balance between glucose influx into the circulation (principally from food and hepatic glucose production) and peripheral glucose clearance. Glucose is stored as glycogen, a 70 kg man typically having a total of 700–1000 g (hydrated) glycogen. Most of this is in the skeletal muscle (400–600 g), and liver (60–125 g), with lesser amounts in other tissues. Glycogen is synthesized from both glucose and the gluconeogenic substrates (see above). Glycogen is a polymeric storage form of glucose. Extension of the polymer by the addition of glucose 6-phosphate subunits is catalysed by glycogen synthase, which is the main regulatory enzyme for glycogen synthesis. This enzyme is itself subject to regulation by a dephosphorylation/phosphorylation cycle controlled by a cyclic AMP dependent protein kinase. Glycogen in skeletal muscle can provide fuel for muscle but does not provide a source of glucose for release into the circulation. Glucose provides approximately 40–60% (on a typical diet in the developed world) of the total fuel expended by the body during a 24 h period. It provides almost all the energy for the central nervous system (especially in well-nourished subjects, although sustained fasting allows cerebral ketone body utilization). During high-intensity exercise and during the 4–6 h postprandial period, glucose is the predominant fuel for the whole body. Glucose is the most efficient fuel for oxidation in terms of the liberation of energy (112.2 kcal or 6 moles ATP per mole of oxygen consumed). Many tissues can use ketone bodies, fatty acids or glucose for their basal metabolism, switching between these different fuels depending upon their availability in the circulation. Glucose is fully oxidized to carbon dioxide and water in skeletal muscle, the brain and the liver. The brain accounts for most of the glucose oxidized in the fasting state (100–125 g/24 h). In the resting, fasted state, skeletal muscle takes up 10–20% of hepatic glucose output: this is not all oxidized but can be converted to lactate, pyruvate, glycerol or the carbon skeletons of amino acids, much of which subsequently return to the liver and act as precursors for gluconeogenesis. Fatty acids (or their partial oxidation products, ketone bodies) are the prime fuel of resting muscle, heart and liver. Other tissues such as red blood cells, skin, adipose tissue and the renal medulla derive energy from glycolysis to lactate and pyruvate, even in the resting state. Glycolysis to lactate is an anaerobic process to which many cells may resort when faced with hypoxia, for example skeletal muscle during high-intensity exercise. The brain can adapt to the utilization of ketone bodies as a source of fuel during a prolonged fast, but this is far from being an instantaneous process, and the more immediate response depends upon increasing glucose supply as a result of the action of the counter-regulatory hormones glucagon, cortisol, catecholamines and growth hormone. Although each can achieve the objective of restoration of blood glucose supply to the brain, the mechanism for each differs. Adrenaline (epinephrine) and glucagon are the ‘rapid response’ team, activating hepatic glucose formation via glycogen phosphorylation, whilst growth hormone and cortisol are the ‘back up’ team inducing the enzymes responsible for gluconeogenesis. Catecholamines can additionally increase circulating non-esterified fatty acid concentrations by upregulating lipolysis so muscle uptake of glucose falls, leaving more glucose for the brain to use. In contrast, if the counter-regulatory hormones are present in excess and there is also a relative deficiency of insulin then hyperglycaemia results; infections and trauma are common causes of increased secretion of catecholamines. Glucose is a hydrophilic molecule that is unable directly to penetrate the lipid bilayer of cells: its uptake into cells is therefore achieved by an energy-independent process of facilitated diffusion down its concentration gradient, mediated by a family of glucose transporter proteins (GLUTs) composed of at least 12 membrane spanning helices with a larger intracellular loop connecting the sixth and seventh helices. These transporters allow uptake of glucose into cells from the interstitial fluid into which glucose diffuses from the bloodstream and, by virtue of differences in kinetics, tissue and subcellular expression profiles, and substrate specificities, mediate specific functions such as glucose sensing (GLUT2) and insulin-dependent glucose uptake (GLUT4) (Table 15.1). The 14 facultative glucose transporters recognized to date are classified into those having high glucose affinity (class I comprising GLUTs 1–4), high fructose affinity (class II, e.g. GLUT5) and novel transporters whose physiology is not yet fully understood (GLUTs 6–14). The different functions of the class I GLUTs are partly predictable from their differing Km values. Glucose transporters 1, 3 and 4 have Km values of ~2–5 mmol/L but GLUT2 has a Km of ~25 mmol/L. This permits high rates of glucose entry into essential cells (e.g. in the central nervous system (CNS), which is relatively protected from neuroglycopaenia by the low Km of its GLUT3 transporters). Pancreatic β-Cells are able to sense increments in blood glucose over a range considerably exceeding normal values via the high Km GLUT2 transporter protein. Certain glucose transporters (GLUT1 and GLUT3) are present on cell surfaces at all times. In contrast, GLUT4 is stored in the cytoplasm when insulin is not present; it responds to insulin by moving from intracellular stores to cell membranes, thereby increasing total transporter number (typically 6–10-fold). When insulin concentrations decline, GLUT4 is removed from cell membranes by endocytosis and rapidly recycled back to its intracellular storage compartments. Erythrocytes are insulin non-responsive because they possess only GLUT1. Many cells can express a variety of different GLUT transporters, and the expression of GLUT receptors changes with circumstances: for example liver cells express more GLUT1 and GLUT3 during starvation. Dysfunction of the insulin-regulated GLUT4 translocation process appears to play a part in the insulin resistance syndrome, and mutations of several transporters (e.g. GLUT1, GLUT2) have been associated with inherited metabolic diseases of carbohydrate metabolism. Tissue glucose uptake via GLUT transporters involves facilitated diffusion down a concentration gradient, the intracellular concentration of glucose being very low because of its active metabolism by phosphorylating enzymes (hexokinases and glucokinases). In resting, postabsorptive subjects, ~ 70% of the body’s glucose metabolism occurs independently of the action of insulin. However, these insulin-independent mechanisms cannot maintain normoglycaemia for very long without insulin orchestrating the response to food and integrating the balance between fatty acid, ketone body and glucose metabolism. Insulin-independent (as well as insulin-dependent) glucose clearance is impaired in subjects with type 2 diabetes and also in normoglycaemic subjects with a family history of diabetes. This suggests that abnormalities in insulin-independent glucose disposal manifest at a very early stage of disease development. This phenomenon of ‘glucose resistance’ appears to be quantitatively important: in normal subjects, as much as half an intravenous glucose load is cleared by non-insulin-mediated glucose disposal. Although polymorphisms of GLUT4 do not appear to be any more common among patients with type 2 diabetes than in the normal population, there is evidence that targeting and trafficking of this glucose transporter may be abnormal in both skeletal muscle and adipose tissue in this condition. The insulin-sensitizing agents metformin and the thiazolidinediones appear to increase cell surface expression of GLUT4, as does physical exercise. The ability to move glucose against a concentration gradient, necessary in the special circumstances of the renal tubules and intestinal epithelium, is conferred by a family of sodium-dependent glucose cotransporters. Sodium-dependent glucose cotransporter 1 (SGLT1) is present in the gut, where it is responsible for absorption of glucose and galactose from the diet. Mutations of SGLT1 are associated with the glucose–galactose malabsorption syndrome that may cause fatal infantile diarrhoea unless these sugars are removed from the diet. Sodium-dependent cotransporter 2 (SGLT2) is a low affinity high capacity transporter that is present in the S1 segment of the proximal renal tubules and is responsible for 90% of renal glucose reabsorption, the remaining glucose being reabsorbed by the high affinity but low capacity SGLT1 transporter found in the S3 segment; reduced function mutations of SGLT2 are associated with renal glycosuria. Drugs that block SGLT2 (thus increasing urinary glucose loss) have recently been licensed in the UK as glucose-lowering agents. Some of the principal intracellular metabolic pathways for glucose, together with their rate-limiting enzymes, are schematically illustrated in Figure 15.2, with the pathways labelled a–k. FIGURE 15.2 Intracellular metabolic pathways for glucose. FFA, free fatty acids; G6PDH, glucose 6-phosphate dehydrogenase; GFAT, glutamine:fructose 6-phosphate amidotransferase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase. Broken lines indicate pathways from which intermediates have been omitted for clarity. Lower case letters refer to reactions, which are discussed in the text. In order to trap glucose within cells (since all the GLUTs are potentially bidirectional), glucose is phosphorylated on entry by a family of hexokinases (a). Hexokinase types I–III are widely expressed and have low Km, but hexokinase type IV (also called glucokinase, and predominantly expressed in liver and the β-Cells of the islets of Langerhans) has a much higher Km (up to 15 mmol/L), permitting it to function as a glucose sensor over (and beyond) the physiological range of blood glucose. Since glucokinase is also the rate-limiting step in glucose metabolism, it thus becomes the ultimate determinant of the rate of insulin secretion from β-Cells. Loss of function mutations of glucokinase are responsible for one form of maturity onset diabetes of the young (MODY 2, which represents approximately 30% of all cases of MODY). Patients present with mildly elevated blood glucose concentrations owing to apparent elevation of the set point for blood glucose concentrations. For reasons that are not totally clear, patients with this condition appear to be at very low risk for the development of diabetic complications and are rarely symptomatic. Although a good response is typically obtained with augmentation of the insulin response by sulfonylurea therapy, this is often not necessary. Although glucokinase might be a potential candidate gene in type 2 diabetes (and a treatment target, given that activating mutations can cause hypoglycaemia in man), mutations appear to be no more common among people with type 2 diabetes than in the general population. Dephosphorylation of glucose (the reverse reaction) is catalysed by glucose 6-phosphatase (b). This process is necessary for the export of glucose (from gluconeogenesis) by hepatic and renal cells in hypo-insulinaemic situations. Deficiency of this enzyme (or of the associated glucose 6-phosphate transport protein) is the cause of glycogen storage disease type 1 (von Gierke disease), and its overactivity is a feature of the increased and relatively insulin-insensitive hepatic glucose production typical of type 2 diabetes. The insulin sensitizing agents metformin and the thiazolidinediones appear to reduce the activity of glucose 6-phosphatase, although it is not clear whether these are direct effects or are mediated through some other upstream action. Having entered the cell and undergone phosphorylation, glucose undergoes one of four distinct metabolic fates. It may be: • metabolized aerobically to yield energy • metabolized anaerobically to yield energy • used to synthesize other molecules (glycerol and the carbon skeletons of certain non-essential amino acids) • stored as glycogen. The dominant flux along each pathway will depend on the tissue (e.g. gluconeogenesis can take place in the liver and kidneys, glycogen storage in liver and muscle, glycerol generation in adipose tissue) and on the prevailing metabolic circumstances such as cellular ATP requirements (requiring glycolysis), ‘housekeeping’ requirements (e.g. plasma membrane stability in erythrocytes), oxygen availability (determining whether glycolysis is anaerobic (yielding lactate) or aerobic (yielding pyruvate, which can be converted to acetyl-CoA and enters the tricarboxylic acid cycle) and alternative substrate availability (e.g. fatty acids or ketone bodies in the fasting state). Some of these pathways, and in particular the rate of glucose oxidation, have been shown to be abnormal in subjects with type 2 diabetes and also in their non-diabetic relatives. Glucose oxidation proceeds via the action of the rate-limiting enzyme phosphofructokinase (c) through various intermediates to pyruvate. Pyruvate may cross the mitochondrial membrane to enter the tricarboxylic acid cycle via the mitochondrial pyruvate dehydrogenase (PDH) enzyme complex (d) (releasing a net 38 molecules of ATP per molecule of glucose) or may remain in the cytosol and be converted anaerobically to lactate via the bidirectional enzyme lactate dehydrogenase (LDH) (e) (yielding only a net two molecules of ATP per molecule of glucose). Pyruvate dehydrogenase in muscle is stimulated by exercise and in most tissues by insulin, but this stimulation is reduced in diabetes. Pyruvate dehydrogenase is inactivated by phosphorylation, catalysed by PDH kinase (which is over expressed in insulin resistance). As a result, glucose metabolism is shifted away from the tricarboxylic acid cycle. This effect may be prevented by activation of peroxisome proliferation activating receptor α (PPARα, see p. 720), one of a family of lipid-activated transcription factors. Mitochondrial diseases have been reported to cause insulin resistance or diabetes (e.g. diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) syndrome, Friedreich ataxia and HIV-associated lipodystrophy); indices of mitochondrial activity (e.g. mitochondrial number and size) are generally reduced in insulin-resistant states. Lactate, generated under anaerobic conditions (and in cells such as erythrocytes, which lack the requisite enzymes) can be used as a substrate for gluconeogenesis either locally or after export to the liver (the Cori cycle), or may enter the tricarboxylic acid cycle after reconversion to pyruvate (f). It has been postulated that an increased availability of substrates such as FFAs or ketone bodies, entering the tricarboxylic acid cycle via acetyl-CoA (g), may lead to excess generation of citrate, with the consequent inhibition of phosphofructokinase (PFK), thus shunting glucose down alternative, non-oxidative pathways (the glucose-FFA or Randle cycle). The consequent reduction in the entry of glucose into the tricarboxylic acid cycle could contribute to diminished glucose oxidation and thus to diabetes. Glycogenolysis (h) is initiated by glycogen phosphorylase, the activity of which is stimulated by glucagon (in liver) and catecholamines (in muscle). In muscle, the glucose liberated from glycogen is used locally as an energy source: it is not exported into the circulation. Muscle glycogen storage via glycogen synthase (i) is diminished in type 2 diabetes, potentially contributing to reduced glucose disposal. Intracellular glucose can also be metabolized through the pentose phosphate shunt (hexose monophosphate shunt) via glucose 6-phosphate dehydrogenase (j). Deficiency of this enzyme causes an X-linked form of haemolytic anaemia, owing to the fact that this pathway is the only route of glucose metabolism in red blood cells; it is prevalent in people originating from the Mediterranean littoral. The rate-limiting enzyme of the hexosamine synthetic pathway (glutamine:fructose 6-phosphate amidotransferase, GFAT) (k) is overactive in diabetes and hyperglycaemia; this pathway leads to the glycosylation of proteins, including transcription factors, and can affect cellular sensing of glucose. The hexosamine pathway is also relevant to the development of complications of diabetes through alterations in endothelial function mediated by endothelial nitric oxide synthase (eNOS), protein kinase A and protein kinase C. Defects in glucose oxidation and glycogen storage may cause shunting of glucose down this pathway, leading to insulin resistance, reduced insulin secretion and diabetic complications. Animal models of glucotoxicity show many of the features of type 2 diabetes, for example reduced GLUT4 translocation (reducing glucose uptake into cells), reduced glycogen synthase activity (reducing glucose incorporation into glycogen), increased hepatic glucokinase activity (increasing intracellular glucose trapping), increased hepatic glucose output and reduced β-Cell glucokinase activity (reducing insulin secretion). Insulin resistance may thus be a defence against excessive intracellular accumulation of glucose, sensed by excessive shunting down the hexosamine synthetic pathway and mediated via the transcriptional regulation of key enzymes and pathways (‘cellular satiety’). Reduced glucose oxidation in pancreatic β-Cells could lead to impairment of insulin secretion, and in other tissues could lead to diminished glucose disposal, both of which are hallmarks of type 2 diabetes. However, the extent to which these abnormalities actually contribute to diabetes rather than relate to specific experimental conditions (e.g. glucose and insulin concentrations), or are simply consequences of some other pathological processes, remains unclear. Insulin is a peptide hormone (51 amino acids arranged in two peptide chains linked by two disulphide bonds, molecular weight 5807 Da), which is secreted by the β-Cells of the islets of Langerhans in the pancreas. In the synthesis of insulin, translation of mRNA yields preproinsulin, a prohormone that undergoes post-translational modification prior to release of the mature insulin molecule. Removal of 24 amino acids from preproinsulin yields proinsulin, which consists of 86 amino acids. Proinsulin is stored in secretory granules prior to release from β-Cells by exocytosis. In healthy subjects, > 90% of proinsulin is converted to mature insulin by the removal of the metabolically inert C-peptide component prior to secretion. The other products of the post-translational modification are either released when exocytosis occurs or are degraded within the secretory granules prior to release. C-peptide is co-secreted in equimolar amounts with mature insulin. In healthy subjects, only small amounts (< 10% of mature insulin output) of proinsulin and partially split proinsulin are released. These ratios are characteristically disturbed in certain pathological states, including autonomous insulin secretion from an insulinoma and type 2 diabetes mellitus. Plasma concentrations of proinsulin and C-peptide are low or undetectable in cases of surreptitious administration of exogenous insulin. Assay of these substances may, therefore, prove helpful in the differential diagnosis of hypoglycaemia in some circumstances. Proinsulin may accumulate in renal failure and its plasma concentration is elevated in familial hyperproinsulinaemia. Substances stimulating the synthesis and storage of insulin include glucose, mannose, leucine, arginine and a variety of metabolizable sugars and sugar derivatives. Most of these also promote secretion. The mechanisms by which insulin release is triggered are the focus of much research. It is apparent that there is an ATP-dependent, sulfonylurea-sensitive K+ channel whose closure is a late event in the intracellular signalling mechanism within the β-Cell; closure triggers calcium influx and exocytosis. It is not clear how this K+ channel is activated, although a wide range of secretagogues can stimulate activation of this final common pathway. The most important of these is hyperglycaemia, although mannose, lactate, arginine, leucine and other amino acids, glucagon, glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic peptide (GIP, known formerly as gastric inhibitory peptide), cholecystokinin, vasoactive intestinal peptide (VIP), sulfonylureas and parasympathetic cholinergic (muscarinic) nerve activity also stimulate insulin release; many of these secretogogues have synergistic effects. Both neural sympathetic tone and circulating catecholamines inhibit insulin secretion. Because of cephalic and gastric influences, oral glucose is a more potent stimulus to insulin secretion than an equivalent amount of intravenous glucose. This difference is known as the ‘incretin’ effect and is predominantly mediated by gut-derived hormones such as the N-terminally truncated GLP-1 (7–36) amide, and GIP. Recently, drugs have become available that augment the release of insulin via the sulfonylurea-sensitive K+ channels of β-Cells (meglitinide analogues) and GLP-1 receptors (GLP-1 analogues), both acting in a glucose-dependent manner. These mechanisms afford the clinically desirable possibility of augmenting insulin release during conditions of hyperglycaemia, while at the same time reducing the risk of hypoglycaemia. They are discussed further in Chapter 16. In healthy adults, insulin is secreted in pulses with a pulse periodicity of 11–15 min. Stimuli of insulin secretion increase the frequency and amplitude of these pulses. Approximately 30–40 U (210–280 nmol) of insulin are secreted per 24 h in healthy subjects of normal weight. Insulin secretion is basal (0.25–1.0 U/h) until glucose concentration exceeds a threshold of ~5 mmol/L, and becomes maximal at a concentration of 15–20 mmol/L. Insulin is secreted into the portal venous system and thus must pass through the liver before reaching the systemic circulation. Approximately half of the insulin is cleared in the ‘first pass’ through the liver. The liver is exposed to insulin concentrations approximately three-fold higher than other tissues during endogenous insulin secretion. The first pass clearance of insulin by the liver is variable, being controlled not only by the prevailing blood glucose concentration but also by factors released from the gut before absorption is complete, and by the integrated action on β-Cell receptors of a range of endocrine and nutritional signals. It is, therefore, not surprising that it is difficult to replicate physiological patterns of insulin concentrations by giving exogenous insulin subcutaneously. Autocrine and paracrine regulation of insulin secretion by pancreatic and gut hormones (which may reach very high concentrations within islets) is not well understood. Increased secretion of insulin involves recruitment of more β-Cells to the secreting mode. Fasting peripheral insulin concentrations vary between 20 and 140 pmol/L (~ 2.9–20 mU/L), as measured by immunoassays in healthy subjects, the higher values being associated with increasing age and obesity. After a typical mixed meal (700–800 kcal), peak plasma insulin concentrations reach ~ 350–580 pmol/L (50–84 mU/L) in lean young adults. The half-life of insulin injected into a peripheral vein is 2–6 min, with most being cleared by the liver and smaller amounts being cleared by other tissues that have insulin receptors, such as skeletal muscle, although there is also non-receptor mediated clearance by a variety of tissue proteases. There are a number of recognized genetic abnormalities of insulin structure involving mutations of the insulin gene and hence altered amino acid sequences. Some of these are listed in Table 15.2. Each of the proinsulin and insulin variants mentioned has reduced biological activity in relation to insulin. This causes a propensity to diabetes, although individuals who can sustain a compensatory hypersecretion may not develop the condition. For example, in familial hyperproinsulinaemia, hypersecretion is usually sufficient to prevent diabetes, whereas in insulin Wakayama, diabetes is usual. There are also recognized associations between some polymorphisms affecting the insulin secretory mechanism (e.g. calpain 10, a molecule that promotes the fusion of secretory granules with cell membranes) and diabetes. TABLE 15.2 Some genetically determined abnormal insulins More common abnormalities of insulin secretion involve loss of the normal pulsatility of insulin release, an early feature of both obesity and type 2 diabetes. The progressive loss of insulin secretory capacity in diabetes is discussed below. Insulin has widespread actions, some of which are listed in Table 15.3. It is the dominant hormone regulating blood glucose concentration. It should be noted that while the mechanisms of its glucoregulatory action have been the subject of extensive research, much less is known about its other actions in health or in insulin-resistant states, although vascular effects (vascular smooth muscle proliferation, vasodilatation), CNS effects (appetite, learning, memory) and effects in relation to growth, differentiation and apoptosis have all been proposed. At present, only two receptors that mediate the actions of insulin have been identified: the insulin receptor and the IGF (insulin-like growth factor) receptor. It is apparent, however, that there are individual dose–response curves for the different actions of insulin in different tissues. For example, the median effective dose (ED50) for insulin’s antilipolytic action on adipose tissue is < 140 pmol/L (20 mU/L) (and may be < 70 pmol/L), while those for inhibition of hepatic glucose output and stimulation of glucose uptake into skeletal muscle are 210–350 pmol/L (30–50 mU/L) and 350–490 pmol/L (50–70 mU/L), respectively. A doubling of insulin concentration inhibits hepatic glucose output by ~80% and stimulates peripheral glucose utilization by ~ 20%. In patients with type 2 diabetes, these differential effects on lipolysis, hepatic glucose output and glucose uptake, coupled with ongoing (if reduced) insulin secretion, are probably responsible for the fact that most individuals are not at risk of developing ketoacidosis (at least for many years), despite the clear defect in glucoregulation that is evident. The different actions of insulin have different time courses, with the glucoregulatory and antilipolytic occurring within a few minutes, and growth regulation and actions dependent upon synthesis of new proteins occurring over periods of hours or days. Intravenous injection of insulin typically has little effect on blood glucose for 5–10 min, the maximal hypoglycaemic action occurring after 5–15 min. Insulin stimulation of skeletal muscle glucose uptake declines with a half-life of 10–20 min after the insulinaemic stimulus has been removed. Proinsulin and partially split proinsulins have metabolic activities generally similar to those of insulin, although their plasma half-lives are 3–5 times longer and their biological potencies are only 8–15% that of insulin. It has been suggested that the hepatic activity of proinsulin may be relatively more potent than its effect on peripheral glucose uptake. The main glucoregulatory effects of insulin are mediated by a transmembrane receptor found on insulin-sensitive cells. This receptor is a glycoprotein, with total molecular weight of 350 000 Da, comprising four peptide chains (two α- and two β-subunits), linked by disulphide bridges (Fig. 15.3). Two isoforms of the receptor (IR-A and IR-D) are formed by alternative splicing. The gene for, and amino acid structure of, the insulin receptor have been characterized and show homology with those of the IGF-1 receptor (see below). Within the intracellular domain of the β-subunit is a tyrosine kinase capability, which is activated when insulin binds with the extracellular domain of the α-subunits. The tyrosine kinase promotes autophosphorylation of the receptor followed by activation of threonine and serine kinases. FIGURE 15.3 The insulin receptor. The insulin receptor gene is located on the short arm of chromosome 19 (19p 13.2). Rare gene mutations have been described, for example leprechaunism and Rabson–Mendenhall syndrome, which result in severe glucose intolerance with resistance to exogenous insulin, and profoundly disordered growth, rather than typical insulin resistance. These conditions are usually lethal in infancy and adolescence, respectively. There are also more common, ‘milder’ polymorphisms of the insulin receptor gene. However, these appear to explain only a small proportion of the marked variance in population insulin sensitivity and they are considered to be a rare (< 1%) cause of type 2 diabetes mellitus. Indeed, most mutations of the insulin gene that have been recognized are not sufficient to cause diabetes alone, although they may render it more likely to occur in the presence of other risk factors. Insulin can have multiple actions, even on a single responsive cell, and hence there are probably several different intracellular pathways mediating its actions. Glucoregulatory and antilipolytic responses are rapid and probably mediated via serine and threonine kinases and cAMP. Stimulation of lipid and protein synthesis, inhibition of proteolysis, the nuclear transcription of RNA and the replication of DNA are slower and have different second messenger systems. Second messenger systems involving diacylglycerol, protein kinase C and glycosyl-phosphatidyl-inositol are under investigation, but abnormalities in them have yet been demonstrated to be necessary or sufficient to cause diabetes in man. As a result of these second messenger cascades, glucose transporter proteins are translocated from within cells to their surface membranes to increase glucose flux into the cytoplasm. After activation, insulin–receptor complexes are internalized by an endocytotic process; receptors are later recycled to the cell surface. Internalization of the insulin is important (and possibly essential) for insulin signals to reach the nucleus and influence cell growth and protein synthesis. Internalization is an important route by which insulin is cleared from the circulation and degraded. The actions of insulin in stimulating DNA transcription and mRNA translation do not depend upon the plasma membrane insulin receptor kinase activity and the second messenger systems discussed above, or on the IGF receptors described below, but rather involve direct effects within the nucleus and on ribosomes. In addition to its acute effects on glucose uptake and release and on lipid metabolism, insulin has growth promoting activity in a variety of tissue culture models. At least two protein hormones, insulin-like growth factors 1 and 2 (IGF-1 and IGF-2), have actions that partly resemble these actions of insulin. The amino acid sequences of these proteins and the base sequences of their coding DNA are known and show some homology with those of insulin. Insulin-like growth factors are weak agonists for the insulin receptor and hence have weak glucoregulatory and antilipolytic effects. In addition, they have growth-promoting effects mediated by two IGF receptors. Insulin itself binds only weakly to IGF receptors. The physiological role of IGFs is not yet fully established. It has been suggested that they act as somatomedins (they were previously called somatomedins C and A, terms that are no longer recommended), in that they are induced by growth hormone and mediate its growth-promoting effects in children. In adults, they are believed to promote the growth of continually dividing cells such as osteoblasts, chondrocytes, fibroblasts and erythroid cells. Adult growth hormone deficiency is manifest by low overall quality of life scores, reduction in lean body mass, centripetal fat distribution, loss of bone mineral, abnormal lipid profile, insulin resistance and changes in the secretion and metabolism of other circulating hormones, for example thyroxine (see p. 367). The effects of growth hormone deficiency on insulin sensitivity are complex. While glucose intolerance is a common feature of acromegaly, in growth hormone deficiency states, adverse changes consequent upon an increase in body fat composition tend to outweigh the lack of a counter-regulatory hormone to produce insulin resistance overall. Replacement therapy in adult growth hormone deficiency produces somewhat unpredictable effects, although a period of increased insulin resistance may herald a modest longer-term improvement. Tumour-related hypoglycaemia may be due to excess production of IGFs, specifically ‘big’ IGF-2. Local and systemic concentrations of IGFs are modulated by a range of binding proteins. While total IGF-2 may not be increased in tumour-related hypoglycaemia, the IGF-2 is present in an immature form which, because of impaired ability to form the normal ternary 150 kD complex with IGF binding protein-3 (IGFBP-3) and the acid-labile subunit (ALS), circulates with greater than normal bioavailability. This effect has been described in a number of tumours, especially those of mesenchymal and epithelial origin. Further discussion of this topic is presented in Chapter 17. It was first observed many years ago that proliferative diabetic retinopathy could regress following hypophysectomy in both animals and humans. This effect appears to be mediated via reduction in IGF-1, owing to the growth hormone deficiency seen after pituitary ablation. It has been hypothesized that the metabolic derangements of diabetes reduce hepatic IGF-1 generation in response to growth hormone (i.e. induce growth hormone resistance), which is then hypersecreted (owing to a lack of negative feedback from IGF-1), reaching concentrations that are able to stimulate IGF-1 generation in non-hepatic tissues. However, studies of pegvisomant, a growth hormone analogue that antagonizes IGF-1 generation in response to growth hormone, have not shown benefit in diabetic retinopathy. Diabetes is the most common metabolic disorder, with a cumulative incidence of 5–10% in people in the developed world aged > 40 years, in whom > 90% of cases are due to type 2 diabetes. Population screening programmes typically reveal that up to half of the subjects found to have type 2 diabetes had previously been undiagnosed. While the burden of type 2 diabetes is increasing exponentially with the epidemic of obesity in many parts of world, the incidence of type 1 diabetes has also been increasing for many years for reasons that are much less apparent. Nevertheless, the prevalence of type 2 diabetes in children is approaching that of type 1 diabetes, having been only ~2–4% of all childhood diabetes prior to 1994, and is predicted to outstrip type 1 diabetes by 2025 on current trends. A high proportion of these children, typically presenting around the time of puberty, require insulin from the time of diagnosis. Whether this is due to the accelerator hypothesis (‘double diabetes effect’), whereby an individual’s risk of contracting type 1 diabetes is increased by the prior existence of type 2 diabetes or insulin resistance, is not clear. Although insulin has many actions, as described above, diabetes is defined only in terms of elevated blood glucose concentrations. Since blood glucose is a continuous variable, the cut-off points for diagnosis are necessarily somewhat arbitrary. Recent changes to the diagnostic criteria for diabetes and glucose intolerance reflect the increased cardiovascular risk evident at even moderate levels of fasting hyperglycaemia (~6.0 mmol/L in some studies). However, the blood glucose threshold for this effect is almost certainly lower than that for the predominantly microvascular complications unique to diabetes mellitus. Some people so diagnosed may not, therefore, necessarily be at risk of developing the unique set of complications (nephropathy, retinopathy, neuropathy) that have traditionally characterized the disease and determined its management. The diagnostic criteria for diabetes mellitus set out by The World Health Organization (WHO) have been adopted by both the American Diabetes Association (ADA) and Diabetes UK, although they differ slightly for glucose intolerance. The WHO introduced diagnostic criteria in 1979, but introduced revised criteria in 2000 to reflect better understanding of ‘milder’ glucose intolerance. The use of glycated haemoglobin (HbA1c) for diagnosis was introduced in 2011. The WHO glucose criteria for diagnosis are shown in Box 15.2 and Table 15.4 and its recommendations regarding the use of HbA1c are shown in Box 15.3. World Health Organization criteria consider fasting and 120 min values in the oral glucose tolerance test (OGTT). The reproducibility of the OGTT leaves much to be desired (the CV of 120 min plasma glucose concentrations is reported to be up to 50%). If a subject fulfils the WHO criteria for diabetes, subsequent improvement of glucose tolerance may occur (e.g. as a result of weight loss or spontaneously), but such individuals are considered to have a lifelong tendency to diabetes. Table 15.4 indicates the criteria for the diagnosis of impaired glucose tolerance (IGT) and impaired fasting glycaemia (IFG), which are metabolic states intermediate between normal glucose tolerance and diabetes. For epidemiological or population screening purposes, the fasting or 2 h value after 75 g oral glucose may be used alone. For clinical purposes, the diagnosis of diabetes should always be confirmed by repeating the test on another day, unless there is unequivocal hyperglycaemia with acute metabolic decompensation or obvious symptoms. Glucose concentrations should not be determined on serum or plasma unless red cells have been immediately removed, or glycolysis will result in an unpredictable underestimation of the true concentrations. It should be stressed that glucose preservatives such as fluoride do not totally prevent glycolysis. If whole blood is used, the sample should be kept at 0–4 °C, and assayed as soon as possible. Subjects with IGT and IFG are at high risk of progression to diabetes; IGT and IFG should therefore be considered as risk categories for future diabetes and/or cardiovascular disease. In certain cases, glycated haemoglobin (HbA1c), the concentration of which reflects glycaemia over several weeks, gives sensitivity and specificity for diagnosis almost equal to that of glucose measurements, and a cut-off value of 48 mmol/mol (6.5%) is now accepted as diagnostic, although it is important to note that a value below this level does not necessarily exclude diabetes. Several classifications of diabetes have been proposed. The most widely used is that proposed by the WHO and adopted by the ADA shown in Box 15.4. However, it should be recognized that unanimity in nomenclature has yet to be achieved, especially in the areas of gestational diabetes, diabetes related to pancreatitis and tropical/malnutrition-related diabetes. BOX 15.4 Aetiological classification of diabetes mellitus I Type 1 diabetes (β-Cell destruction, usually leading to absolute insulin deficiency)
Glucose metabolism and the pathophysiology of diabetes mellitus
PHYSIOLOGY AND PATHOPHYSIOLOGY OF GLUCOSE HOMOEOSTASIS
Introduction: the maintenance of normoglycaemia
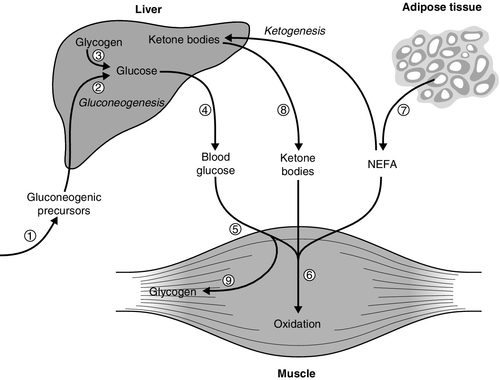
 reduces the production of gluconeogenic precursors such as glycerol, alanine and lactate,
reduces the production of gluconeogenic precursors such as glycerol, alanine and lactate,  reduces activity of hepatic gluconeogenic enzymes and
reduces activity of hepatic gluconeogenic enzymes and  increases hepatic glycogenolysis to glucose. These effects contribute to
increases hepatic glycogenolysis to glucose. These effects contribute to  reduced hepatic glucose output. Insulin
reduced hepatic glucose output. Insulin  increases cellular glucose uptake mediated by insulin-sensitive glucose transporters (GLUT4) and
increases cellular glucose uptake mediated by insulin-sensitive glucose transporters (GLUT4) and  reduces competition for glucose oxidation by alternative fuels (‘Randle effect’). The reduction of competing fuels involves
reduces competition for glucose oxidation by alternative fuels (‘Randle effect’). The reduction of competing fuels involves  inhibition of NEFA release from adipose tissue and
inhibition of NEFA release from adipose tissue and  reduced hepatic ketogenesis.
reduced hepatic ketogenesis.  Insulin promotes glucose storage as glycogen.
Insulin promotes glucose storage as glycogen.
Normal glucose metabolism
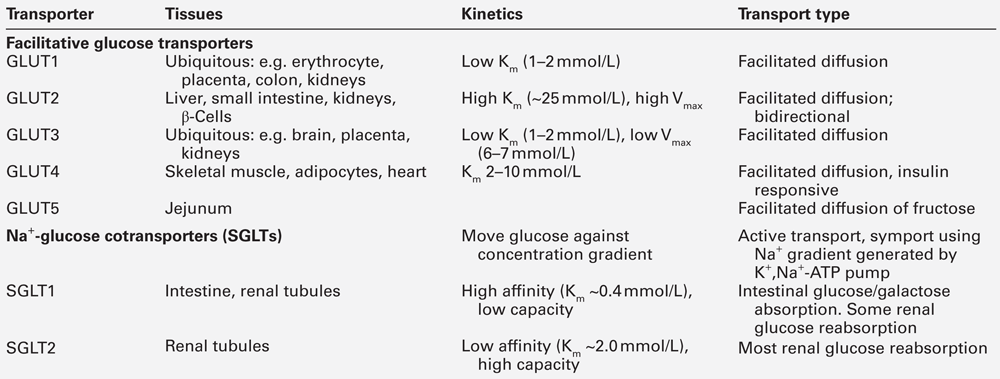
Glucose transporters
The fate of intracellular glucose and its role in diabetes
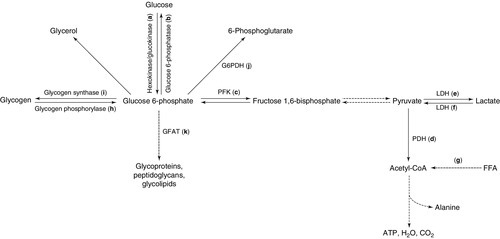
Insulin
Biosynthesis
Secretion and pharmacokinetics
Abnormalities of the synthesis and secretion of insulin
Condition
Abnormality
Consequences
Familial hyperproinsulinaemia
Failure to cleave
C-peptide from proinsulin
True insulin
concentrations and glucose tolerance normal
Insulin Chicago
Leu for Phe substitution at position 25 of β chain
Reduced receptor binding
Insulin Los Angeles
Ser for Phe substitution at position 24 of β chain
Reduced receptor binding
Insulin Wakayama
Leu for Val substitution at position 3 of α chain
Reduced receptor binding
Proinsulins Boston and Tokyo
His for Arg substitution at position 65
Inability to cleave proinsulin to insulin
Proinsulin Providence
Asp for His substitution at position 10 of β chain
Inability to cleave proinsulin to insulin
Proinsulin Kyoto
Leu for Arg substitution at position 65
Inability to cleave proinsulin to insulin
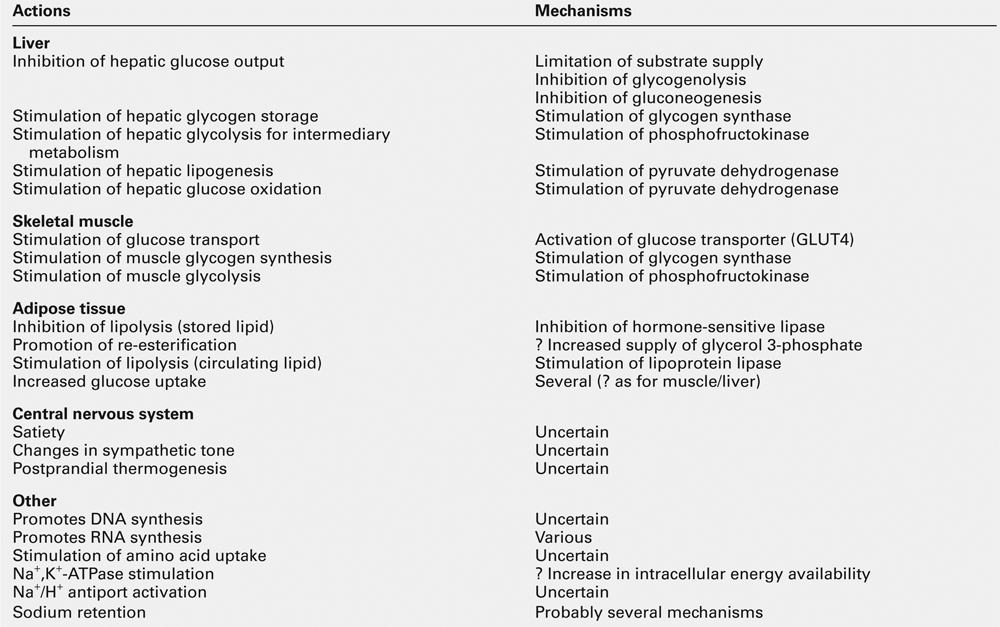
Actions of insulin
The insulin receptor
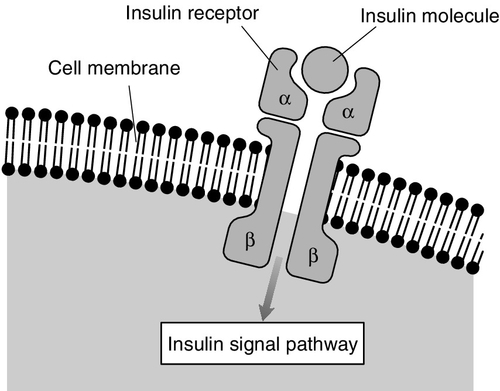
Second messengers mediating the effects of insulin
Insulin-like growth factors and their receptors
CLASSIFICATION AND DIAGNOSIS OF DIABETES MELLITUS
Introduction
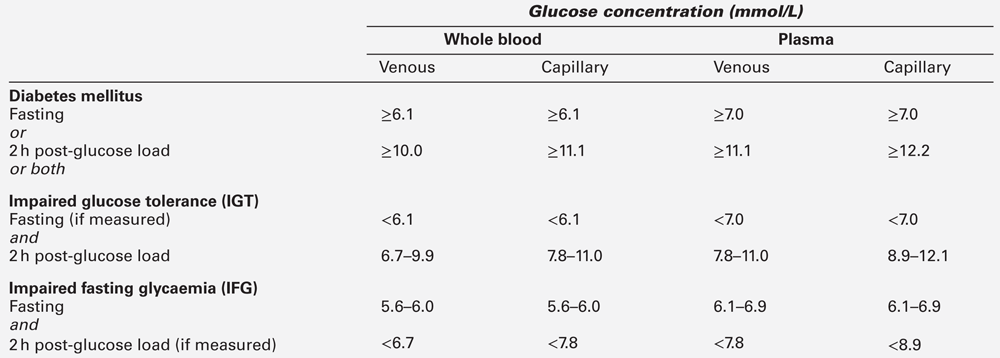
Definitions
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine