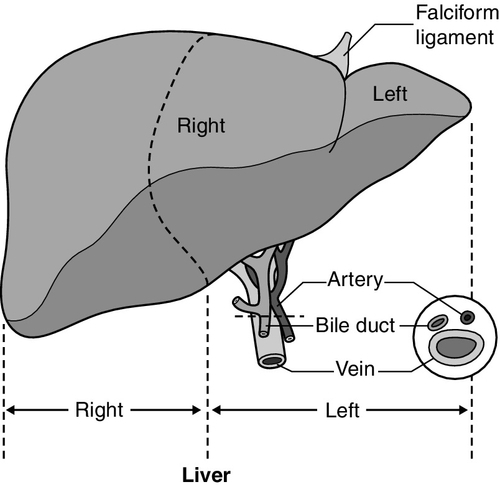
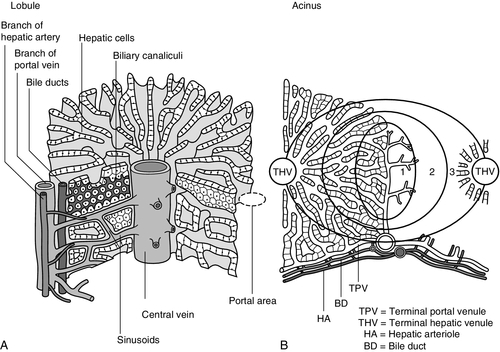
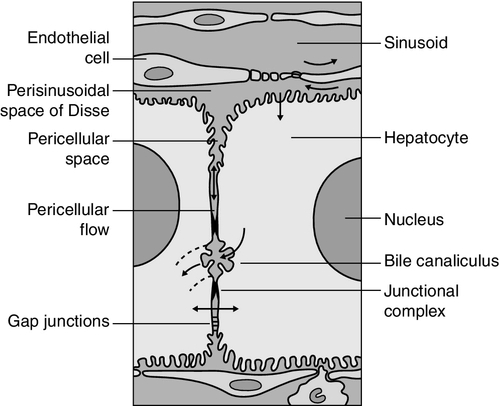
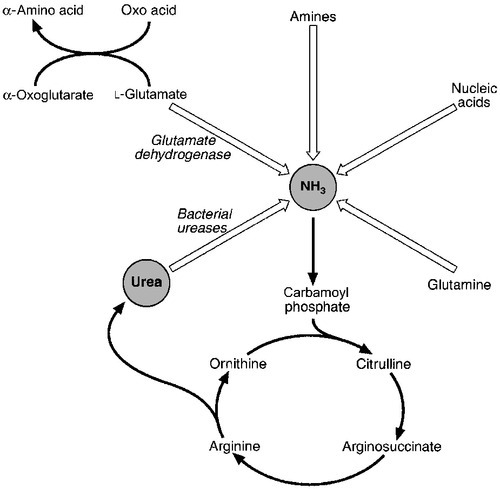
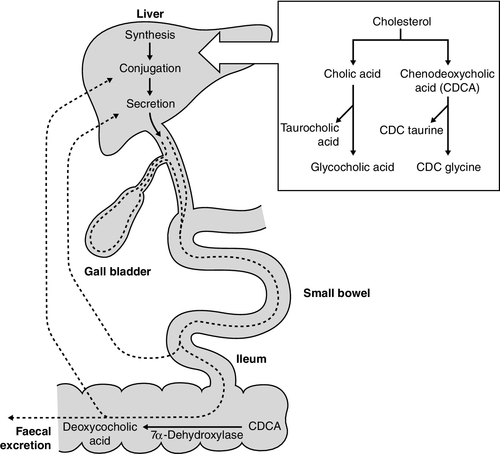
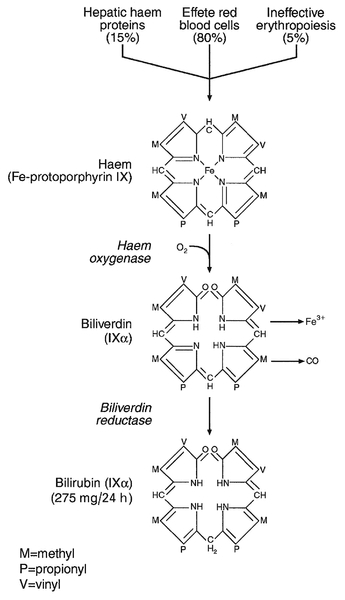
CHAPTER 13 Roy A. Sherwood; Adrian Bomford CHAPTER OUTLINE Bile, bile ducts and biliary drainage Biotransformation and excretion Bilirubin and bile pigment metabolism Quantitative evaluation of liver function Differential diagnosis of jaundice The inherited hyperbilirubinaemias Monitoring response to therapy ABNORMAL LIVER FUNCTION TESTS IN ASYMPTOMATIC PATIENTS NORMAL LIVER FUNCTION TESTS IN THE PRESENCE OF OVERT LIVER DISEASE ROLE OF LIVER FUNCTION TESTS IN ASSESSING PROGNOSIS It is widely accepted that the term ‘liver function tests’, as currently used in clinical practice, is a misnomer. Certainly this is true as compared with pulmonary or renal function tests. These describe the measurement of distinct physiological and biochemical organ functions that have meaning in the absence of any organ pathology. In marked contrast, most of the parameters that comprise the standard liver ‘function’ tests, such as the plasma activities of certain aminotransferases, which have major and well-defined roles within the cell, have no functional significance at all in plasma, where they are simply markers of hepatocyte disruption. Their measurement is only of any significance when applied to liver pathology. To this extent, understanding of the conventional liver function tests relies on a broad grasp of the principles of liver disease, and the present chapter should, therefore, be read in conjunction with Chapter 14. This is not to say that the function of the liver is not well understood. But, while it is conventional to list the functions that the liver can perform, this detracts from gaining a broad conceptual picture of what the liver ‘does’. Essentially, the liver is a regulatory barrier between the systemic circulation and the organism’s environment experienced via the gut. The job of the acinus, the functional unit of the liver, is to regulate the concentrations of solutes entering the systemic circulation via the terminal hepatic venules or being excreted in the bile. It is the principal organ of metabolic homoeostasis, that is, maintenance of blood composition within physiologically acceptable limits by the conversion, synthesis and release of components required by other organs and by removal of toxic substances that may be injurious to tissues. This chapter reviews briefly the anatomy, physiology and biochemistry of the normal liver as a basis for understanding the tests currently applied in clinical practice and those that may be developed in the future. The macroscopic and microscopic anatomy of the liver is difficult to understand, partly because of its inherently complicated three-dimensional structure and partly because of the recent trend to replace simple (but misleading) morphological descriptions with more accurate, but less obvious, functional descriptions. The liver has a dual blood supply. Arterial blood, direct from the aorta, is supplied via the hepatic artery from the coeliac axis. The second source is the portal vein, which is formed by the joining of the superior mesenteric and splenic veins and which collects blood from the gut. After passage through the sinusoids (see below), blood drains from the liver via the hepatic veins at the posterior aspect of the liver into the inferior vena cava and thence to the right side of the heart. The portal venous system delivers about 80% of the blood and 20% of the oxygen supplied to the liver. A major cause of abnormal function in chronic liver disease is disturbance of blood flow through the liver, consequent on the fibrosis that follows chronic liver cell damage, but if the portal vein is blocked, relatively normal liver function can be maintained for many years. Arterial occlusion occurring suddenly, for example as a result of trauma, leads to acute liver failure, but more gradual occlusion by a tumour mass is compatible with normal liver function for quite long periods. The liver is a wedge-shaped organ located in the right upper quadrant of the abdomen. Its mass varies with that of the individual, being in the order of 22 g/kg body weight. In a typical 70 kg subject, the liver weighs about 1.5 kg. It has a large right lobe, a smaller left lobe anteriorly and two further small lobes, the quadrate and the caudate lobes. These lobes relate to the venous drainage, not to the portal distribution (see below). Thus, the left hepatic vein drains the left hepatic lobe and the right and middle hepatic veins drain the right hepatic lobe. In terms of the portal structures, there are two functional lobes defined by the right and left portal veins. The division is marked by a line joining the inferior vena cava and the gallbladder bed (Fig. 13.1). FIGURE 13.1 The anterior surface of the liver. The labelling in the upper part of the figure represents the conventional description into a right and left lobe separated by the falciform ligament. The lower part of the labelling refers to the right and left lobes defined by distribution of the portal structures. The right and left portal structures (portal vein, hepatic artery and bile ducts) enter the functional right and left lobes, respectively. Hepatocytes, the hepatic parenchymal cells, comprise about 80% of the total cell mass of the liver. As viewed under the microscope, the functional unit of the liver appears to be the acinus, often termed the lobule, and it is in terms of this structure that pathological changes are described. Since liver biopsy has, under many circumstances, become the de facto ‘gold standard’ of liver disease diagnosis, it is important to understand this terminology. It is also important to understand that this unit does not comprise a homogeneous collection of hepatocytes and that there is marked functional heterogeneity across the acinus, with different zones having different physiological and biochemical functions (see below). The acinar/lobular concept of the structure of the liver is based on a central efferent terminal hepatic venule surrounded by radially oriented plates of hepatocytes and sinusoidal channels. Three to five portal tracts (or ‘triads’), containing branches of the portal vein, hepatic artery and bile duct, are situated around the periphery of each acinus (Fig. 13.2A). FIGURE 13.2 (A) The conventional hepatic acinus (lobule) based on the central vein with surrounding portal tracts. (B) The hepatic acinus and its three functional zones. The axis is formed by the terminal portal venule, hepatic arteriole and bile duct. Blood flows from the periphery of the acinus to the terminal hepatic venule. The acini represent microcirculatory units comprising groups of liver cells, each fed by a single terminal portal venule and hepatic arteriole, the blood from which passes via the sinusoids into a terminal hepatic vein (Fig. 13.2B). Flow is unidirectional, from portal tract to hepatic vein. The 20 or so hepatocytes that separate the portal tracts from the terminal hepatic vein in an acinus have been arbitrarily separated into three zones, through which the portal blood passes sequentially. Zone 1 is conventionally used to describe hepatocytes nearest the afferent arteriole in the portal tract (the periportal area), and those surrounding the terminal hepatic vein, in the ‘centrilobular’ or ‘perivenular’ area, are described as zone 3. Between these two zones is an ill-defined area (zone 2), which is intermediate in terms of the composition of the perfusing blood. It must be emphasized that there are no anatomical boundaries between these three zones but, clearly, as blood perfuses each zone sequentially, its composition is altered and this leads to the functional heterogeneity of hepatocytes across the acini in response to the changes in their microenvironment. Zone 1 hepatocytes receive blood rich in oxygen and have a high level of metabolic activity. It is not surprising, therefore, that oxidative functions of the liver tend to be performed mainly by hepatocytes in zone 1, with metabolic processes that can operate at lower oxygen tensions occurring mainly in zone 3. A detailed discussion of the functional heterogeneity of the three zones is not undertaken here, but we mention some examples that could be important if damage to the liver is not uniform. Thus, oxidative metabolism (the respiratory chain, citric acid cycle and fatty acid oxidation), gluconeogenesis, urea synthesis and the production and excretion of bile all occur mainly in zone 1, while glycolysis, glutamine synthesis from ammonia, and xenobiotic metabolism occur predominantly in zone 3. It is not difficult to appreciate that different pathological insults may differentially damage the various zones. Perhaps blood tests may eventually be developed to investigate the integrity of the different zones and thereby reduce the need for histological examination in the diagnosis of different liver disorders. Hepatocytes are arranged in single-cell sheets or ‘plates’, supported by a fine mesh of a collagenous material (reticulin) and separated from overlying fenestrated endothelial lining cells by the space of Disse (Fig. 13.3). The hepatocytes are exposed to blood flowing through the sinusoids on each side of the plates. Their sinusoidal surfaces have a microvillar structure that greatly increases the surface area of the cell membrane, thereby facilitating efficient exchange of solutes between the blood and the cells. Bile produced by the hepatocytes is excreted via specific transporters located in the membrane of the biliary canaliculi (see below). The latter are formed by invaginations of the smooth basolateral membranes between adjacent hepatocytes, and also have a microvillar structure. Direct communication between the blood and bile is prevented by tight junctions in the basolateral membranes on either side of the biliary canaliculi. In addition to the endothelial cells that filter fluid exchanged between the sinusoidal lumen and the space of Disse, the sinusoids also contain Kupffer cells (a major part of the reticuloendothelial system), hepatic stellate cells (vitamin A-storing, fat-storing cells, also known as Ito cells, that produce several connective tissue components) and liver-resident natural killer cells with antitumour activity. These non-parenchymal cells are an area of intensive research; current evidence suggests that they play a major role in synthesis of growth factors responsible for control of liver regeneration and development of fibrosis in liver disorders (see below). FIGURE 13.3 Ultrastructure of the liver. Solutes can pass out of the sinusoid, which does not have a conventional basement membrane, and across the space of Disse. Here they may be taken up across the hepatocyte membrane and subsequently across the canalicular membrane, or enter the canaliculus through the ‘paracellular pathway’ via the intercellular junctions. The biliary canaliculi formed between adjacent hepatocytes are continuous with canaliculi between other hepatocytes within the plane of the liver cell plates, and eventually drain into ductules lined by specific biliary epithelial cells and ultimately into the major bile ducts, thence to the common bile duct and the gut. Interruption of the flow of bile, the exocrine secretion of the liver, is responsible for many of the signs of hepatobiliary disease. Bile pigment and bile acid metabolism are described below, and the pathological anatomy of the liver and biliary tract is described in Chapter 14. The fact that the liver has a great capacity for regeneration is the rationale for much of the practice of clinical hepatology, particularly for undertaking prolonged periods of liver- intensive care during acute liver failure and for the feasibility of extensive resection, particularly for the management of primary and, less commonly, secondary malignant liver tumours. Hepatic regeneration has been studied mainly in animals, most extensively the rodent model, after partial hepatic resection. Twenty-four hours after liver resection, there is extensive division of the remaining cells accompanied by a surge in DNA synthesis; this is followed 24 h later by increased replication of the non-parenchymal cells. The initiation of these events is by growth factors that are now being identified. Epidermal growth factor (EGF), transforming growth factor α (TGFα) and hepatocyte growth factor (HGF) are all involved in switching on regeneration, and transforming growth factor β (TGFβ) is involved in switching it off. Normally quiescent differentiated hepatocytes replicate rapidly after resection, while intrahepatic precursor cells, termed oval cells, proliferate and generate lineage only in situations where hepatocyte proliferation is blocked or delayed. Bone marrow cells can generate oval cells, but such transdifferentiation is now known to be inefficient and occurs only rarely. Because the various functions of the liver may fail at different times, creating differing clinical pictures, it is necessary to consider each function separately. The physiology and biochemistry of the liver subsume most of intermediary metabolism and, as such, are clearly beyond the scope of this chapter. Other than giving a brief outline, emphasis is placed on those functions whose measurement or disturbance is important in liver tests or pathology, respectively. As has been known for many years, removal of the liver in an animal leads to death from hypoglycaemia. A major metabolic function of the liver is to store sugar and reduce the variations in blood glucose consequent upon the human habit of eating intermittently. Thus, during a meal, the liver stores glucose as glycogen and then releases it (glycogenolysis) slowly when food is not being eaten. This is particularly important for those organs such as the brain and red blood cells that have an obligatory requirement for glucose. Specific glucose transporter molecules located in the sinusoidal membrane that mediate facilitated diffusion are involved in both processes. Between meals, as the supply of glycogen decreases (only about 75 g can be stored), the liver starts to make glucose (gluconeogenesis) from other sources, particularly lactate, but also pyruvate, glycerol and alanine, although only from alanine is there a significant net production of glucose. During more prolonged periods of starvation, the total body requirement for glucose falls and energy demand is increasingly met by production of ketone bodies, derived mainly from fatty acids via acetyl-CoA. Apart from being stored, glucose is used by the liver as an energy substrate via glycolysis and the citric acid cycle, or for the synthesis of fatty acids and triglycerides. Insulin is secreted in response to the rise in blood glucose concentration after a meal and promotes an increase in peripheral glucose uptake and a decrease in gluconeogenesis. In acute liver failure, the liver may not be able to maintain an adequate concentration of blood glucose and hypoglycaemia may become a life-threatening complication; in chronic liver disease, hyperglycaemia is more common, most likely because of a failure of the liver to store glycogen and failure of peripheral tissues to take up glucose adequately. The liver also metabolizes other dietary sugars including fructose and galactose, converting them to glucose phosphates (see Chapter 14). After a meal, dietary triglycerides are hydrolysed to free fatty acids and monoglycerides by pancreatic lipases and dissolved in an aqueous medium, facilitated by the detergent action of bile salts excreted by the liver into the gut. The liver meets its own metabolic energy requirements, and those of the body as a whole, by mitochondrial β-oxidation of short chain fatty acids. The resultant acetyl-CoA either enters the citric acid cycle or reacts with another molecule of acetyl-CoA to form ketone bodies. Although the role of the liver is central to the oxidation of fatty acids, most tissues contain the enzymes required to undertake complete oxidation. The liver also synthesizes fatty acids, triglycerides, cholesterol, phospholipids and lipoproteins. Disturbances of fatty acid metabolism, including decreased oxidation (as in excessive alcohol consumption), increased hepatic fatty acid synthesis and decreased breakdown of triglycerides to fatty acids and glycerol, may all be involved in the development of fatty liver (‘steatosis’), an increasingly prevalent problem found in obesity and as an additional or incidental histological finding in many liver disorders. Hepatic protein metabolism is central to the assessment of liver function, and its disturbance underlies many of the clinical complications that occur in severe liver cell dysfunction. Other than the immunoglobulins, most circulating proteins are synthesized wholly or largely by the liver, and the concentrations of several are used as a measure of hepatic synthetic function. Apart from albumin, transcobalamin II and C-reactive protein, all are glycoproteins. Glycosylation (often with terminal sialic acid residues on the carbohydrate moieties) has several functions. In some instances (e.g. fibronectin), it serves to make the protein resistant to proteolysis; in others it affects function. In yet others (e.g. caeruloplasmin), it affects the half-life of the protein in the blood, because hepatocytes possess receptors that can bind galactose and some other carbohydrate residues exposed after removal of sialic acid and remove the desialylated glycoproteins from the circulation. Disturbance in glycosylation produces some specific defects in protein structure that may be useful clinically in the diagnosis of alcoholic (see p. 242) and malignant liver disease (see Chapter 14). A 70 kg man on a normal diet needs to excrete between 10 and 20 g of nitrogen per 24 h. This derives, in the form of ammonia, from amino acids that are surplus to requirements (and cannot be stored) and those that are not reutilized after normal turnover. The ammonia is converted into urea in the liver and excreted by the kidneys. The liver processes dietary amino acids arriving via the portal vein and from breakdown of muscle proteins, both for its own requirements and for export to peripheral tissues. Aromatic amino acids (AAA: phenylalanine, tyrosine and tryptophan) are metabolized by the liver, but hepatic extraction of branched chain amino acids (BCAA: leucine, isoleucine and valine) is small and these are taken up largely by muscle. The ratio of BCAA/AAA is decreased in acute liver failure and this alteration forms the basis of one theory of the pathogenesis of hepatic encephalopathy, namely that it is due to the toxic effects of increased concentrations of ammonia on the brain. The major pathways of ammonia production and clearance are shown in Figure 13.4. Amino acids first undergo transamination to glutamate, followed by oxidative deamination with the formation of ammonia. The resultant ammonia is fed into the Krebs–Henseleit (urea) cycle and excreted as urea or stored transiently as glutamine (through the action of glutamine synthase). Additional ammonia is produced by the action of intestinal bacteria on dietary protein and urea present in gut contents. Plasma ammonia concentration is measured in many laboratories and a raised concentration is taken as evidence that an encephalopathic state is due to hepatic pathology. Measurement of the various enzymes involved in the urea cycle is increasingly undertaken for diagnosis of inherited abnormalities of urea synthesis. FIGURE 13.4 Production and disposal of ammonia. The lungs and kidneys are effective in excreting volatile and water-soluble substances, respectively, but many compounds, both exogenous (such as drugs) and endogenous, including end products of metabolism, are lipid soluble and non-volatile. Many of these compounds are toxic, and a vital function of the liver is to render such substances more water soluble so that they can be excreted in urine or bile. Two phases of biotransformation of metabolites by the liver are recognized. In phase I, a suitable polar group is made available, which is conjugated in phase II. Phase I reactions occur in the smooth endoplasmic reticulum and are mediated mainly by the mixed function oxidase system (cytochrome P450 isoenzymes) that utilize atmospheric oxygen, typically generating hydroxylated or carboxylated compounds. Phase II reactions involve their subsequent conjugation by the action of glucuronyl transferases with glucuronic acid, acetyl or methyl radicals or, in the case of bile acids, with glycine, taurine or sulphate (see below). There is considerable functional heterogeneity of the glucuronyl transferases, of which there are several isoenzymes that have varied substrate specificities, particularly for exogenous compounds. An alternative, non-oxidative pathway for biotransformation of both endogenous and exogenous substances is by conjugation with reduced glutathione by glutathione S-transferases. The constituents of bile, the exocrine secretion of the liver, are conjugated bile salts, cholesterol, phospholipids (mainly lecithin), bilirubin mono- and diglucuronides, electrolytes and small amounts of protein. The liver is the major site of cholesterol biosynthesis and the sole site of conversion of cholesterol into bile acids, which are the major organic anions excreted by the liver. The primary bile acids, cholic and chenodeoxycholic acid, are conjugated with either glycine or taurine to form bile salts, which enhances their solubility at the pH of body fluids (Fig. 13.5). This facilitates their main function of solubilizing both biliary cholesterol and the products of dietary fat digestion resulting from lipid hydrolysis. Secondary bile acids, deoxycholic and lithocholic acids, are derived from primary bile acids by the action of intestinal bacterial 7α-dehydroxylase, either as bile salts or deconjugated bile acids. Most of the bile acids reaching the gut are reabsorbed in the terminal ileum and return via the portal vein to the liver (enterohepatic circulation) where, as free acids, they are again conjugated and excreted. A third metabolite, ursodeoxycholate (a stereoisomer of chenodeoxycholic acid), is found in trace amounts and has been classified as a tertiary bile acid. This enterohepatic circulation is regulated by the activities of distinct bile salt transport proteins, including the canalicular bile salt export pump, the ileal sodium-dependent bile salt transporter and the hepatic sinusoidal sodium–taurocholate co-transporting polypeptide. Several other bile salt transporters and organic anion-transporting polypeptides have been characterized (see below). During bile secretory failure (cholestasis), bile salt transport proteins undergo adaptive responses that serve to protect the liver from retention of toxic bile salts and facilitate non-hepatic routes of bile salt excretion. The measurement of serum bile acids has been extensively investigated as a test of liver function (see p. 243). FIGURE 13.5 Synthesis and enterohepatic circulation of bile acids. Liver function tests have four potential applications. 2. As an aid to making a specific diagnosis. While functional tests are clearly distinct from diagnostic tests, it is still reasonable to assume that certain patterns of dysfunction may be characteristic of particular diseases. 3. To establish the severity of liver dysfunction or damage once a specific diagnosis has been established. This is important from a prognostic point of view, although the standard biochemical ‘liver function tests’ do not always reflect accurately the severity of tissue damage. 4. To monitor the progression of the disease and any response to therapeutic intervention. Within this framework, two classes of tests will be considered. The first are the standard liver function tests – a group of tests often applied irrespectively of the suspected diagnosis and to which all the caveats about lack of true functional assessment apply. The second group includes those biochemical assays used for assessment of liver disease in specific situations, for example α1-antitrypsin in suspected deficiency of this protein and α-fetoprotein in suspected primary hepatic cancer (hepatocellular cancer). The latter are described briefly here, for the sake of completeness, and in more detail in the next chapter in the context of their diagnostic application. The standard liver function tests are usually considered to include the plasma total bilirubin concentration, the activities in plasma of certain enzymes (particularly alanine and/or aspartate aminotransferases, alkaline phosphatase and γ-glutamyltransferase), and the total plasma protein, albumin and globulin concentrations. The prothrombin time (discussed later) also provides useful information about the synthetic capacity of the liver in the short term, as it is a functional measure of factor VII (among others) concentration and this clotting factor has a short half-life in plasma (see below). It has been estimated that this group of tests will correctly allocate patients to a liver disease/non-liver disease category in about 75% of cases. Although bilirubin has been characterized as a non-toxic metabolic product of a relatively minor metabolic pathway, marked elevation in its plasma concentration leads to the alarming sign of jaundice and usually suggests the presence of underlying liver or biliary tract disease that may range from trivial to life-threatening, especially in neonates (see below). Accurate interpretation of the laboratory tests associated with bile pigment metabolism in the jaundiced individual requires a clear understanding of the physiology and biochemistry of the bile pigments. Most bilirubin is derived from the breakdown of haem, itself derived from senescent red blood cells (Fig. 13.6). A much smaller proportion comes from other haemoproteins such as catalase, myoglobin and the cytochromes. An even smaller fraction comes from ‘ineffective erythropoiesis’, although this may represent a significant source of bilirubin in haematological conditions such as thalassaemia and pernicious anaemia. The initial and rate-limiting step is the oxidation of haem to biliverdin by haem oxygenase; this is followed by reduction (catalysed by biliverdin reductase) to bilirubin, with the production of an equimolar amount of carbon monoxide and ferric (Fe3 +) iron. These reactions take place in the macrophages of the reticuloendothelial system, predominantly in the liver, spleen and bone marrow. The resultant ‘unconjugated’ bilirubin is tightly bound to albumin in a 1:1 molar ratio, but additional binding sites of lower affinity are recruited in hyperbilirubinaemic states. This binding limits extrahepatic uptake of the potentially toxic unconjugated bilirubin and facilitates transport to the liver. Other molecules, such as thyroxine and certain drugs, can compete for albumin binding sites and thereby displace bilirubin, although the clinical relevance of this displacement is limited, except possibly in neonates. FIGURE 13.6 Formation of bilirubin. Bilirubin, tightly bound to albumin, is actively transported across the hepatocyte sinusoidal membrane and binds to ligandin (glutathione transferase B). It is then conjugated with glucuronic acid by the action of uridine diphosphate (UDP)-glucuronosyltransferase (glucuronyl transferase) to form mono- and diglucuronides and thereby rendered water soluble (Fig. 13.7). Glucuronidated bilirubin is secreted via an active transport mechanism into the biliary canaliculi and thence reaches the gut. The components of this transport pathway have been identified as products of genes encoding multidrug resistance (MDR) proteins (MRPs) and MDR-associated proteins. The protein MDR3 is known to transport phosphatidylcholine to chaperone bile salts, while MRP2 pumps glucuronidated compounds, that is, conjugated bilirubin as well as organic anions. Secretion is probably rate limiting in the overall transport of bilirubin from plasma to bile. In the gut, some bilirubin is deconjugated by bacterial glucuronidases and (being fat soluble again) is reabsorbed, but most is oxidized to urobilinogen; this is further metabolized to other pigments, particularly stercobilin, and excreted (Fig. 13.8
Assessment of hepatic function and investigation of jaundice
INTRODUCTION
ANATOMY OF THE LIVER
The hepatic circulation
Macroscopic structure
Microscopic structure
The acinus
Ultrastructure
Bile, bile ducts and biliary drainage
HEPATIC REGENERATION
PHYSIOLOGICAL FUNCTIONS
Carbohydrate metabolism
Lipid metabolism
Protein metabolism
Synthesis
Metabolism of amino acids and disposal of urea
Biotransformation and excretion
Bile secretion
LIVER FUNCTION TESTS
Bilirubin and bile pigment metabolism
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


