INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Draw a simple scheme outlining the key biochemical reactions involving cobalamin (vitamin B12) and folate.
Understand how cobalamin and folate are absorbed from the gut and transported in plasma.
Explain the mechanisms that cause megaloblastic morphology in the bone marrow and blood and describe these features.
List the causes of cobalamin deficiency.
List the causes of folate deficiency.
Hematopoiesis depends upon orderly cell division for the logarithmic expansion of progenitor cells into large numbers of circulating blood cells. In the megaloblastic anemias, DNA synthesis is impaired, leading to slowing or arrest of cellular division during the DNA synthesis phase of the cell cycle (S phase). A high fraction of cells suffering from such defects undergo programmed cell death (apoptosis). In the bone marrow, the decreased survival of hematopoietic progenitors leads to reduced production of circulating cells (ineffective hematopoiesis). Because RNA synthesis and cytoplasmic differentiation are relatively unaffected, progenitors and progeny that survive are enlarged (macrocytic). The main cause of megaloblastic anemias is deficiency of either cobalamin (vitamin B12) or folic acid, vitamins that are essential for DNA replication and repair. In addition, chemotherapeutic drugs that inhibit DNA synthesis can result in findings similar to those seen in cobalamin or folate deficiency. It is not surprising that the clinical phenotype extends to other tissues that rely on continuous and robust cellular proliferation and differentiation, particularly the gastrointestinal tract.
Understanding the pathophysiology of the megaloblastic anemias requires knowledge about the absorption, transport, and utilization of folate and cobalamin as well as familiarity with the key chemical reactions in which these vitamins are essential cofactors.
PHYSIOLOGY OF COBALAMIN AND FOLATE
Cobalamin is a complex organic molecule consisting of a tetrapyrole corrin ring, similar in structure to heme except that the divalent metal atom in the center of the ring is cobalt rather than iron. Like heme iron, the cobalt atom in the corrin ring binds to two axial ligands. One is a benzimidazole nucleotide, whereas the other can be either a methyl group (methylcobalamin) or an adenosyl group (adenosylcobalamin). Cobalamin is found in all foods of animal origin including meat, fish, and dairy products. Food cobalamin is tightly bound to proteins. Following ingestion, some cobalamin in food is transferred to human haptocorrin in saliva. As depicted in Figure 6-1, the acidic environment of the stomach enables efficient release and transfer of the remaining food cobalamin to haptocorrin in gastric juice. After transit to the duodenum, the increase in pH enables the transfer of cobalamin from haptocorrin to intrinsic factor, a transport protein secreted by gastric parietal cells. The cobalamin-intrinsic factor complex resists digestion and travels down the gut until it encounters epithelial cells in the distal ileum that express cubilin, a receptor with specificity for this bimolecular complex. The cobalamin that is absorbed in the ileum exits the basolateral side of the mucosal epithelial cell into the plasma where it traverses the portal circulation into the liver. Here cobalamin binds transcobalamin, a plasma transport protein that is functionally analogous to transferrin, the transport protein for iron. As in iron homeostasis, the liver is the principal storage site for cobalamin. The uptake of the circulating transcobalamin-cobalamin complex by receptors on plasma membranes is the principal and probably the only way that the vitamin is taken into cells other than liver cells. In keeping with the broad importance of cobalamin for the biochemical reactions described below, transcobalamin receptors are expressed on a wide variety of cells.
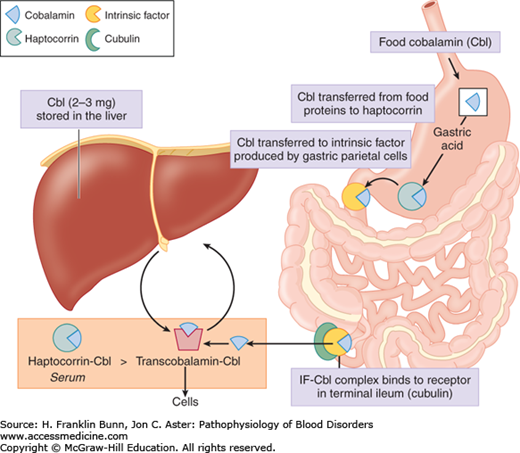
The transcobalamin-cobalamin complex makes up only a minority of the cobalamin in plasma. About 70% to 90% is bound to haptocorrin, derived primarily from leukocytes of the myeloid lineage. Haptocorrin is secreted into a number of bodily fluids including saliva and gastric juice, as mentioned earlier. Its biologic function is unclear.
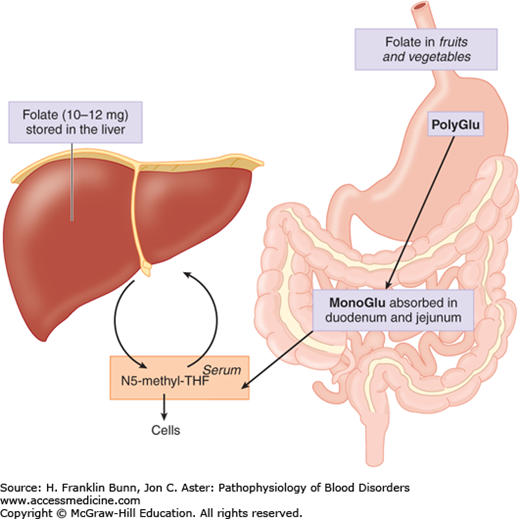
Folic acid, commonly designated folate, refers to a group of compounds consisting of a tricyclic pteroyl group linked to one or more glutamic acid residues. Food sources include fruits, vegetables, liver, and meat, in all of which folate exists primarily as polyglutamate conjugates. The bioactivity of folate in food may be attenuated or destroyed by prolonged cooking. As shown in Figure 6-2, during transit in the gut, folate polyglutamates are hydrolyzed to the monoglutamate derivative, which is readily absorbed by a specific transmembrane channel into the enterocytes of the duodenum and jejunum. It is further metabolized in enterocytes into N5-methyl tetrahydrofolate (N5-methyl THF), which exits these cells and circulates freely in the plasma. Several types of folate receptors have been identified on cell surfaces, but their role in mediating uptake has not been explained. Like cobalamin and iron, folate is stored mainly in the liver.
BIOCHEMISTRY OF COBALAMIN AND FOLATE
These cofactors participate in a series of reactions involved in the biosynthesis of a variety of products including amino acids, metabolic intermediates, purines, and nucleotides. As summarized in Figure 6-3, the two forms of cobalamin mentioned earlier participate in different reactions.
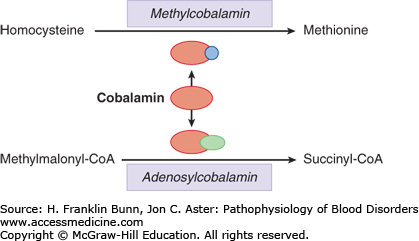
Adenosylcobalamin is required for the conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA. As discussed later in this chapter, measurement of methylmalonate in the serum is a useful test for identifying individuals with cobalamin deficiency. As shown in Figures 6-3 and 6-4, methylcobalamin is required for the conversion of homocysteine to methionine.
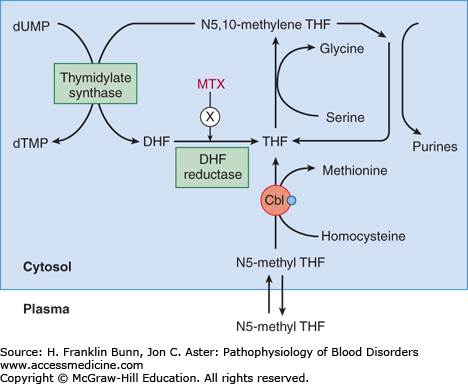
After N5-methyl THF enters the cell from the plasma, it is conjugated to polyglutamate, which prevents egress back into the plasma. Transfer of the methyl group from N5-methyl THF to methionine results in the formation and maintenance of adequate intracellular levels of THF. This reaction exemplifies the primary function of folate as a conduit for the addition of one-carbon moieties such as methyl and formyl groups to organic compounds. The conversion of the amino acid serine to glycine enables the transfer of a methyl group to THF to form N5,10-methylene THF, which is then used for both de novo purine biosynthesis and for the conversion of deoxyuridylate to deoxythymidylate. Thus cobalamin and folate play critical roles in production of the building blocks for DNA synthesis. As discussed in detail in the next section, deficiency in either of these two vitamins results in impairment of cell division, with adverse consequences particularly in proliferating cells such as in the bone marrow and gastrointestinal epithelium.
PATHOPHYSIOLOGY AND FEATURES OF MEGALOBLASTIC ANEMIA
The impairment in DNA synthesis imposed by cobalamin or folate deficiency results in striking morphologic abnormalities in the bone marrow and peripheral blood, often referred to as megaloblastic changes. The bone marrow reveals increased cellularity and hyperplasia of erythroid precursors. The model diagram at the bottom of Figure 6-5 compares the maturation of normal and megaloblastic erythroid cells. Throughout the stages of erythroid development, the megaloblastic cells are larger. Although cytoplasmic differentiation, as assessed by increasing hemoglobin production, is normal, nuclear maturation is retarded, a phenomenon called nuclear-cytoplasmic dyssynchrony
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


