Drug Metabolism
COPING WITH EXPOSURE TO XENOBIOTICS
Substances foreign to the body, or xenobiotics, are metabolized by the same enzymatic pathways and transport systems that are used for normal metabolism of dietary constituents. Drugs are considered xenobiotics and most are extensively metabolized in humans. The capacity to metabolize xenobiotics has made development of drugs very time consuming and costly due in large part to:
• Interindividual variations in the capacity of humans to metabolize drugs
• Drug-drug interactions
• Metabolic activation of chemicals to toxic and carcinogenic derivatives
• Species differences in expression of enzymes that metabolize drugs, thereby limiting the use of animal models to predict effects in humans
Most xenobiotics are subjected to 1 or multiple enzymatic pathways that constitute phase 1 oxidation and phase 2 conjugation. Metabolism serves to convert these hydrophobic chemicals into more hydrophilic derivatives that can easily be eliminated from the body through the urine or the bile.
Many drugs are hydrophobic, a property that allows entry through the lipid bilayers into cells where the agents can interact with their target receptors or proteins. This property of hydrophobicity renders drugs difficult to eliminate, because in the absence of metabolism, they accumulate in fat and cellular phospholipid bilayers in cells. The xenobiotic-metabolizing enzymes convert drugs and other xenobiotics into derivatives that are more hydrophilic and thus easily eliminated through excretion into the aqueous compartments of the tissues.
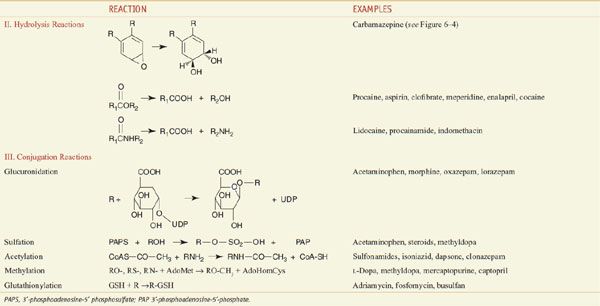
Drug metabolism that leads to elimination also plays a major role in diminishing the biological activity of a drug. For example, (S)-phenytoin, an anticonvulsant used in the treatment of epilepsy, is virtually insoluble in water. Metabolism by the phase 1 cytochromes P450 (CYPs) followed by phase 2 uridine diphosphate-glucuronosyltransferases (UGTs) produces a metabolite that is highly water soluble and readily eliminated from the body (Figure 6–1). Metabolism also terminates the biological activity of the drug.
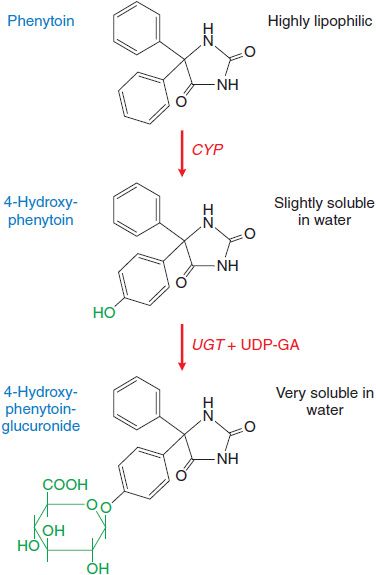
Figure 6–1 Metabolism of phenytoin by phase 1 cytochrome P450 (CYP) and phase 2 uridine diphosphate-glucuronosyltransferase (UGT). CYP facilitates 4-hydroxylation of phenytoin. The hydroxy group serves as a substrate for UGT that conjugates a molecule of glucuronic acid (in green) using UDP-glucuronic acid (UDP-GA) as a cofactor. This converts a very hydrophobic molecule to a larger hydrophilic derivative that is eliminated via the bile.
Paradoxically, these drug metabolizing enzymes can also convert certain chemicals to highly reactive, toxic, and carcinogenic metabolites or carcinogens. Depending on the structure of the chemical substrate, xenobiotic-metabolizing enzymes can produce electrophilic metabolites that react with nucleophilic cellular macromolecules such as DNA, RNA, and protein. Reaction of these electrophiles with DNA can sometimes result in cancer through the mutation of genes, such as oncogenes or tumor suppressor genes. This potential for carcinogenic activity makes testing the safety of drug candidates of vital importance, particularly for drugs that will be used chronically.
THE PHASES OF DRUG METABOLISM
Xenobiotic metabolizing enzymes are grouped into those that carry out: phase 1 reactions, which include oxidation, reduction, or hydrolytic reactions; and the phase 2 reactions, in which enzymes catalyze the conjugation of the substrate (the phase 1 product) with a second molecule (Table 6–1). Oxidation by phase 1 enzymes adds or exposes a functional group, permitting the products of phase 1 metabolism to serve as substrates for the phase 2 conjugating or synthetic enzymes. While many phase 1 reactions result in the biological inactivation of the drug, phase 2 reactions produce a metabolite with improved water solubility, thereby facilitating drug elimination. The example of phase 1 and phase 2 metabolism of phenytoin is shown in Figure 6–1.
Table 6–1
Xenobiotic Metabolizing Enzymes
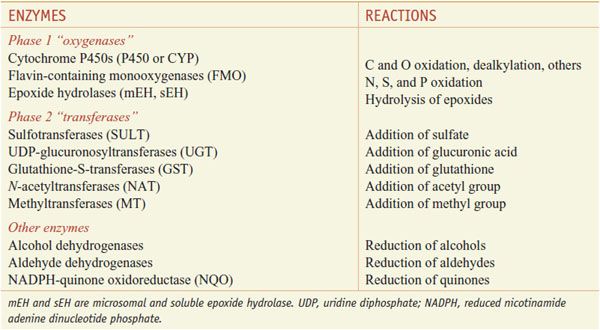
The phase 1 enzymes lead to the introduction of functional groups such as –OH, –COOH, –SH, –O–, or NH2. The phase 1 oxidation reactions are carried out by CYPs, flavin-containing monooxygenases (FMOs), and epoxide hydrolases (EHs).
CYPs and FMOs comprise superfamilies of enzymes containing multiple genes. The addition of functional groups does little to increase the water solubility of the drug, but can dramatically alter the biological properties of the drug. Reactions carried out by phase 1 enzymes usually lead to the inactivation of a drug. However, in certain instances, metabolism, usually the hydrolysis of an ester or amide linkage, results in bioactivation of a drug. Inactive drugs that undergo metabolism to an active drug are called prodrugs. For example, the antitumor drug cyclophosphamide, is bioactivated to a cell-killing electrophilic derivative (see Chapter 61); clofibrate, used to reduce triglyceride levels, is converted in the cell from an ester to an active acidic metabolite.
Phase 2 enzymes facilitate the elimination of drugs and the inactivation of electrophilic and potentially toxic metabolites produced by oxidation. Phase 2 enzymes include several superfamilies of conjugating enzymes, such as the glutathione-S-transferases (GSTs), UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), N-acetyltransferases (NATs), and methyltransferases (MTs). These conjugation reactions usually require the substrate to have oxygen (hydroxyl or epoxide groups), nitrogen, or sulfur atoms that serve as acceptor sites for a hydrophilic moiety, such as glutathione, glucuronic acid, sulfate, or an acetyl group.
In the case of the UGTs, glucuronic acid is delivered to the functional group, forming a glucuronide metabolite that is more water soluble and is targeted for excretion either in the urine or bile. When the substrate is a drug, these reactions usually convert the original drug to a form that is not able to bind to its target receptor, thus attenuating the biological response to the drug.
SITES OF DRUG METABOLISM
Xenobiotic metabolizing enzymes are found in most tissues in the body, with the highest levels located in the GI tract (liver, small and large intestines). The liver is the major “metabolic clearing house” for both endogenous chemicals (e.g., cholesterol, steroid hormones, fatty acids, and proteins) and xenobiotics. The small intestine plays a crucial role in drug metabolism because drugs that are orally administered are absorbed by the gut and taken to the liver through the portal vein. The xenobiotic-metabolizing enzymes located in the epithelial cells of the GI tract are responsible for the initial metabolic processing of most oral medications. The absorbed drug then enters the portal circulation for its first pass through the liver, where it can undergo significant metabolism. A portion of active drug escapes metabolism in the GI tract and liver and enters the systemic circulation; subsequent passes through the liver result in more metabolism of the parent drug until the agent is eliminated. Thus, drugs that are poorly metabolized remain in the body for longer periods of time (have longer elimination half-lives). Other organs that contain significant xenobiotic-metabolizing enzymes include tissues of the nasal mucosa and lung, which play important roles in the metabolism of drugs that are administered as aerosols.
At the cellular level, the phase 1 CYPs, FMOs, and EHs, and some phase 2 conjugating enzymes, notably the UGTs, are all located in the endoplasmic reticulum of the cell (Figure 6–2). Once subjected to oxidation, drugs can be directly conjugated by the UGTs (in the lumen of the endoplasmic reticulum) or by the cytosolic transferases such as GST and SULT. The metabolites can then be transported out of the cell into the bloodstream. Hepatocytes, which constitute >90% of the cells in the liver, carry out most drug metabolism and produce conjugated substrates that can also be transported through the bile canalicular membrane into the bile, from which they are eliminated into the gut (see Chapter 5).
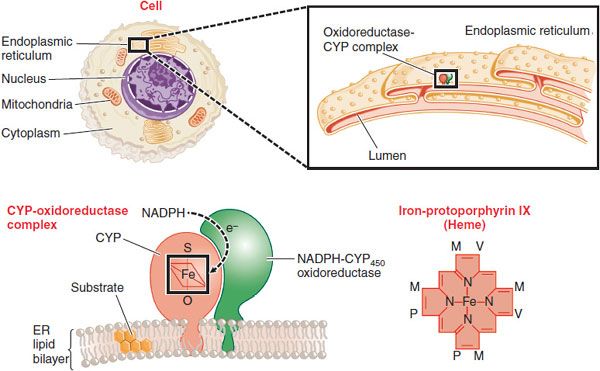
Figure 6–2 Location of CYPs in the cell. The figure shows increasingly microscopic levels of detail, sequentially expanding the areas within the black boxes. CYPs are embedded in the phospholipid bilayer of the endoplasmic reticulum (ER). Most of the enzyme is located on the cytosolic surface of the ER. A second enzyme, NADPH-cytochrome P450 oxidoreductase, transfers electrons to the CYP where it can, in the presence of O2, oxidize xenobiotic substrates, many of which are hydrophobic and dissolved in the ER. A single NADPH-CYP oxidoreductase species transfers electrons to all CYP isoforms in the ER. Each CYP contains a molecule of iron-protoporphyrin IX that functions to bind and activate O2. Substituents on the porphyrin ring are methyl (M), propionyl (P), and vinyl (V) groups.
PHASE 1 REACTIONS
CYPs: THE CYTOCHROME P450 SUPERFAMILY
The CYPs are a superfamily of enzymes, all of which contain a molecule of heme that is noncovalently bound to the polypeptide chain (see Figure 6–2). The heme iron binds oxygen in the CYP active site, where oxidation of the substrates occurs. The H+ is supplied through the enzyme NADPH-cytochrome P450 oxidoreductase and its cofactor NADPH. Metabolism of a substrate by a CYP consumes 1 molecule of O2 and produces an oxidized substrate and a molecule of water. Depending on the nature of the substrate, the reaction is “uncoupled,” consuming more O2 than substrate metabolized and producing what is called activated oxygen or O2–. The O2– is usually converted to water by the enzyme superoxide dismutase. Elevated O2–, a reactive oxygen species (ROS), can give rise to oxidative stress that is detrimental to cells and associated with diseases.
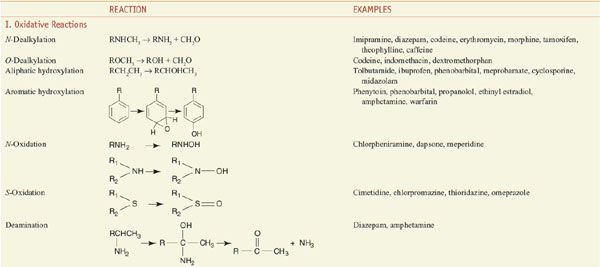
Among the diverse reactions carried out by mammalian CYPs are N-dealkylation, O-dealkylation, aromatic hydroxylation, N-oxidation, S-oxidation, deamination, and dehalogenation (Table 6–2). CYPs are involved in the metabolism of dietary and xenobiotic chemicals, in the synthesis of endogenous compounds (e.g., steroids; fatty acid-derived signaling molecules, such as epoxyeicosatrienoic acids), and in the production of bile acids from cholesterol. In contrast to the drug-metabolizing CYPs, the CYPs that catalyze steroid and bile acid synthesis have very specific substrate preferences. For example, the CYP that produces estrogen from testosterone, CYP19 or aromatase, can metabolize only testosterone or androstenedione and does not metabolize xenobiotics. Specific inhibitors for aromatase, such as anastrozole, have been developed for use in the treatment of estrogen-dependent tumors (see Chapters 40 and 60–63). CYPs involved in bile acid production have strict substrate requirements and do not participate in xenobiotic or drug metabolism.
Table 6–2
Major Reactions Involved in Drug Metabolism
The CYPs that carry out xenobiotic metabolism can metabolize structurally diverse chemicals. This is due both to multiple forms of CYPs, to the capacity of a single CYP to metabolize many structurally distinct chemicals, to significant overlapping substrate specificity amongst CYPs, and to the capacity of CYPs to metabolize a single compound at different positions on the molecule. Indeed, CYPs are promiscuous in their capacity to bind and metabolize multiple substrates (see Table 6–2). This property sacrifices metabolic turnover rates; CYPs metabolize substrates at a fraction of the rate of more typical enzymes involved in intermediary metabolism and mitochondrial electron transfer. As a result, drugs have, in general, half-lives in the range of 2-30 h, while endogenous compounds have half-lives of the order of seconds or minutes (e.g., dopamine and insulin).
The extensive overlapping substrate specificities by the CYPs is one of the underlying reasons for the predominance of drug-drug interactions. When 2 coadministered drugs are both metabolized by a single CYP, they compete for binding to the enzyme’s active site. This can result in the inhibition of metabolism of 1 or both of the drugs, leading to elevated plasma levels. If there is a narrow therapeutic index for the drugs, the elevated serum levels may elicit unwanted toxicities. Drug-drug interactions are among the leading causes of adverse drug reactions (ADRs).
THE NAMING OF CYPs. Genome sequencing has revealed the existence of 57 putatively functional genes and 58 pseudogenes in humans. These genes are grouped, based on amino acid sequence similarity, into a superfamily composed of families and subfamilies with increasing sequence similarity. CYPs are named with the root CYP followed by a number designating the family, a letter denoting the subfamily, and another number designating the CYP form. Thus, CYP3A4 is family 3, subfamily A, and gene number 4.
A DOZEN CYPs SUFFICE FOR METABOLISM OF MOST DRUGS. In humans, 12 CYPs (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5) are known to be important for metabolism of xenobiotics. The liver contains the greatest abundance of xenobiotic-metabolizing CYPs, thus ensuring efficient first-pass metabolism of drugs. CYPs are also expressed throughout the GI tract, and in lower amounts in lung, kidney, and even in the CNS. The most active CYPs for drug metabolism are those in the CYP2C, 2D, and 3A subfamilies. CYP3A4, the most abundantly expressed in liver, is involved in the metabolism of over 50% of clinically used drugs (Figure 6–3A). The CYP1A, 1B, 2A, 2B, and 2E subfamilies are not significantly involved in the metabolism of therapeutic drugs, but they do catalyze the metabolic activation of many protoxins and procarcinogens.
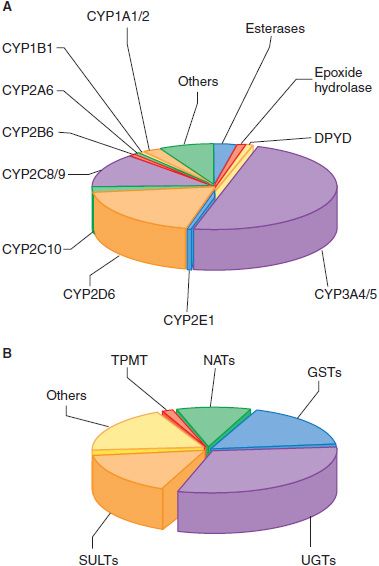
Figure 6–3 The fraction of clinically used drugs metabolized by the major phase 1 and phase 2 enzymes. The relative size of each pie section represents the estimated percentage of drugs metabolized by the major phase 1 (panel A) and phase 2 (panel B) enzymes, based on studies in the literature. In some cases, more than a single enzyme is responsible for metabolism of a single drug. CYP, cytochrome P450; DPYD, dihydropyrimidine dehydrogenase; GST, glutathione-S-transferase; NAT, N-acetyltransferase; SULT, sulfotransferase, TPMT, thiopurine methyltransferase; UGT, UDP-glucuronosyltransferase.
CYPs AND DRUG-DRUG INTERACTIONS. Differences in the rate of metabolism of a drug can be caused by drug interactions. Most commonly, an interaction occurs when 2 drugs (e.g., a statin and a macrolide antibiotic or antifungal agent) are metabolized by the same enzyme and affect each other’s metabolism. Thus, it is important to determine the identity of the CYP that metabolizes a particular drug and to avoid coadministering drugs that are metabolized by the same enzyme. Some drugs can also inhibit CYPs independently of being substrates for a CYP.
For example, the common antifungal agent, ketoconazole (NIZORAL), is a potent inhibitor of CYP3A4 and other CYPs, and coadministration of ketoconazole with an anti-HIV viral protease inhibitor reduces the clearance of the protease inhibitor and increases its plasma concentration and the risk of toxicity. For most drugs, information found on the package insert lists the CYP that metabolizes the drug and determines the potential for drug interactions.
Some drugs are CYP inducers that can increase not only their own rates of metabolism, but also induce metabolism of other coadministered drugs (see below and Figure 6–8).
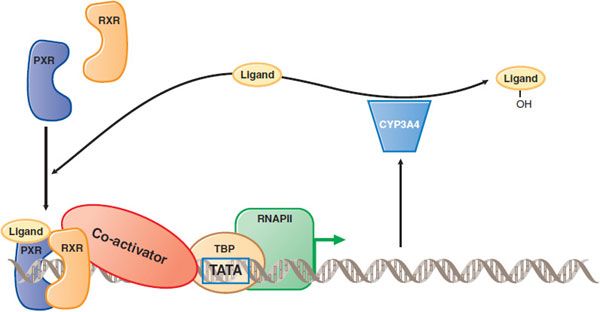
Figure 6–8 Induction of drug metabolism by nuclear receptor–mediated signal transduction. When a drug such as atorvastatin (the Ligand) enters the cell, it can bind to a nuclear receptor such as the pregnane X receptor (PXR). PXR then forms a complex with the retinoid X receptor (RXR), binds to DNA upstream of target genes, recruits coactivator (which binds to the TATA box binding protein, TBP), and activates transcription by RNA polymerase II (RNAP II). Among PXR target genes are CYP3A4, which can metabolize the atorvastatin and decrease its cellular concentration. Thus, atorvastatin induces its own metabolism. Atorvastatin undergoes both ortho and para hydroxylation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


