55
Cancer: An Overview
Robert K. Murray, MD, PhD, Molly Jacob, MB BS, MD, PhD, & Joe Varghese, MB BS, MD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Present an overview of important aspects of the biochemical and genetic features of cancer cells.
Present an overview of important aspects of the biochemical and genetic features of cancer cells.
![]() Describe important properties of oncogenes and tumor suppressor genes.
Describe important properties of oncogenes and tumor suppressor genes.
![]() Briefly describe the concepts of genomic instability, aneuploidy, and angiogenesis in tumors.
Briefly describe the concepts of genomic instability, aneuploidy, and angiogenesis in tumors.
![]() Discuss the use of tumor markers for following responses to treatments and to detect recurrences.
Discuss the use of tumor markers for following responses to treatments and to detect recurrences.
![]() Appreciate that recent understanding of the biology of cancer has led to the development of various new therapies.
Appreciate that recent understanding of the biology of cancer has led to the development of various new therapies.
BIOMEDICAL IMPORTANCE
Cancers constitute the second most common cause of death, after cardiovascular disease, in the USA and many other countries. Approximately 6-7 million people around the world die from cancer each year, and this figure is projected to increase. Humans of all ages develop cancer, and a wide variety of organs are affected. Worldwide, the main types of cancer accounting for mortality are those involving the lung, stomach, colon, rectum, liver and breast. Other types of cancers that lead to death include cervical, esophageal, and prostate cancers. Skin cancers are very common, but apart from melanomas, are generally not as aggressive as those mentioned above. The incidence of many cancers increases with age. Hence, as people live longer many more will develop the disease. Hereditary factors play a role in some types of tumors. Apart from great individual suffering caused by the disease, the economic burden to society is immense.
SOME GENERAL COMMENTS ON NEOPLASMS
A neoplasm refers to any abnormal new growth of tissue. It may be benign or malignant in nature. The term “cancer” is usually associated with malignant tumors. Tumors can arise in any organ in the body and result in different clinical features, depending on the location of the growth.
Cancer cells are characterized by certain key properties: (1) they proliferate rapidly and display diminished growth control, (2) they display loss of contact inhibition in vitro, and (3) they invade local tissues and spread (metastasize) to other parts of the body. These properties are characteristic of cells of malignant tumors. It is the last property that is generally responsible for the deaths of patients who have cancer. Cells of benign tumors also show diminished control of growth, but do not invade local tissue or spread to other parts of the body. Other important properties of cancer cells are as follows: (1) they are self-sufficient in growth signals, (2) they are insensitive to anti-growth signals, (3) they stimulate local angio-genesis, and (4) they are often able to evade apoptosis. These points are summarized in Figure 55–1.
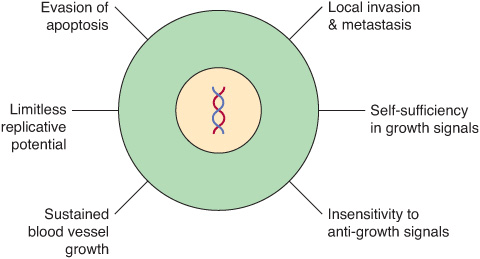
FIGURE 55–1 Six major features of cancer cells. Other important properties of cancer cells are shown in Figure 55–2. (With permission, After Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell 2000;100:57).
Figure 55–2 shows a number of other important properties associated with cancer cells. These various points will be discussed below.
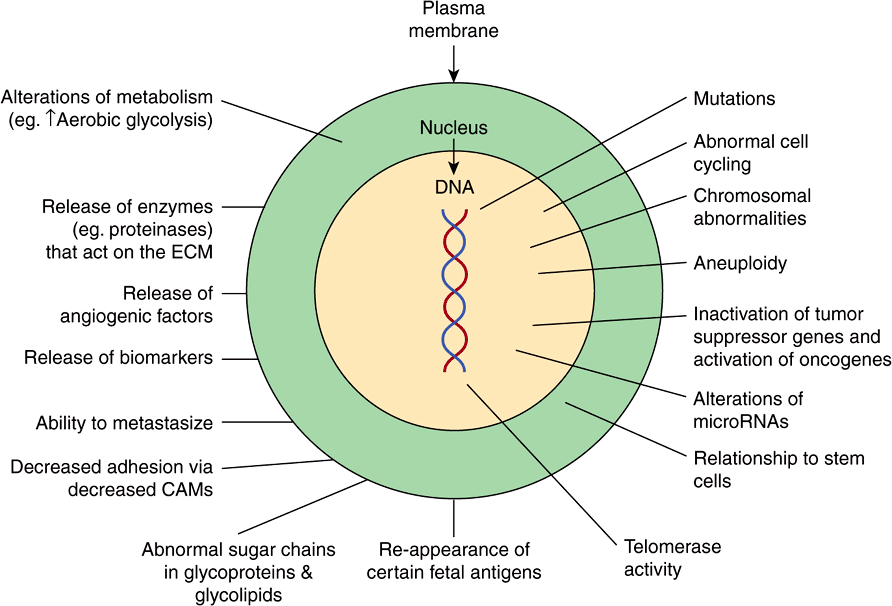
FIGURE 55–2 Some biochemical and genetic changes occurring in human cancer cells. Many changes, in addition to those indicated in Figure 55–1, are observed in cancer cells, only some of which are shown here. The roles of mutations in activating oncogenes and inactivating tumor suppressor genes are discussed in the text. Abnormalities of cell cycling and of chromosome structure, including aneuploidy, are common. Alterations of microRNA molecules that regulate gene activities have been reported, and the relationship of stem cells to cancer cells is a very active area of research. Telomerase activity is often detectable in cancer cells. Tumors sometimes synthesize certain fetal antigens, which may be measurable in the blood. Changes in plasma membrane constituents (eg, alteration of the sugar chains of various glycoproteins—some of which are cell adhesion molecules—and glycolipids) have been detected in many studies, and may be of importance in relation to decreased cell adhesion and metastasis. Various molecules can pass out of cancer cells and can be detected in the blood as tumor biomarkers. Angiogenic factors and various proteinases are also released by some tumors. Many changes in metabolism have been observed; for example, cancer cells often exhibit a high rate of aerobic glycolysis. (CAM, cell adhesion molecule; ECM, extracellular matrix.)
The central issues in cancer are to elucidate the biochemical and genetic mechanisms that underlie the uncontrolled growth of cancer cells, their ability to invade and metastasize and to develop successful treatments that destroy cancer cells, while causing minimal damage to normal cells. Considerable progress has been made in understanding the basic nature of cancer cells, with a central finding being that cancer is a disease due to abnormalities in key genes. However, many aspects of the behavior of cancer cells, in particular their ability to spread, have yet to be fully explained. In addition, despite improvements in treatment of certain types of cancers, therapies are still often unsuccessful. The study of cancer (oncology) is a huge area, so this short chapter can only introduce the reader to some key concepts.
A Glossary at the end of this chapter summarizes the meanings of many of the terms used.
FUNDAMENTAL FEATURES OF CARCINOGENESIS
Nonlethal genetic damage is the initiating event in carcinogenesis. There are principally four classes of genes, which when affected by such damage, can result in the development of a tumor. These are proto-oncogenes, tumor suppressor genes, genes involved in DNA repair, and those that are involved in apoptosis. Cancer is of clonal origin, with a single abnormal cell multiplying to become a mass of cells forming a tumor. Carcinogenesis is thus a multistep process, with multiple genetic alterations occurring in cells, transforming normal cells into malignant ones. Hence, a tumor often takes several years to develop.
CAUSES OF GENETIC DAMAGE
Genetic damage can be due to acquired or inherited mutations. The former occur due to exposure to environmental carcinogens while the latter are hereditary. Such hereditary abnormalities result in a number of familial conditions that predispose to hereditary cancer. These mutations are found in specific genes (eg, tumor suppressor genes) present in the germ cells and are discussed later.
Spontaneous mutations, some of which may predispose to cancer, occur at a frequency of approximately 10-7 to 10-6 per cell per generation. This rate will increase in tissues subject to a high rate of proliferation, increasing the generation of cancer cells from affected parent cells. Oxidative stress (see Chapter 45), by producing increased numbers of reactive oxygen species, may be a factor in increasing the mutation rate.
RADIANT ENERGY, CHEMICALS, AND CERTAIN VIRUSES ARE THE MAJOR KNOWN CAUSES OF CANCER
In general, there are three classes of carcinogens, exposure to which result in tumor formation. These are radiant energy, chemicals, and certain oncogenic viruses (see Figure 55–3). The first two cause mutations in DNA, and the third class generally acts by introducing novel genes into normal cells.
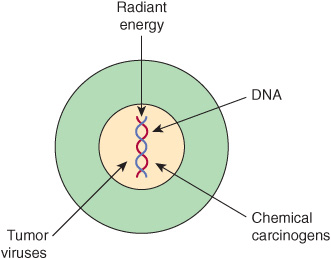
FIGURE 55–3 Radiant energy, chemical carcinogens and certain viruses can cause cancer.
We shall only describe briefly how radiant energy, chemicals, and oncogenic viruses cause cancer.
Radiant Energy can be Carcinogenic
Ultraviolet rays, x-rays, and γ-rays are mutagenic and carcinogenic. Extensive studies have shown that these agents can damage DNA in a number of ways, including the lesions listed in Table 55-1. Mutations in DNA, due to such damage, are thought to be the basic mechanism of carcinogenicity caused by radiant energy although the exact pathways are still under investigation. X-rays and γ-rays can cause formation of reactive oxygen species (ROS), which can also be mutagenic and probably contribute to the carcinogenic effects of radiant energy.
TABLE 55–1 Some Types of DNA Damage Caused by Radiant Energy

Exposure to ultraviolet radiation is common due to exposure to sunlight, which is its main source. Ample evidence exists to show that such radiation is linked to cancers of the skin. The risk of developing a skin cancer due to ultraviolet radiation increases with increasing frequency and intensity of exposure and decreasing melanin content of skin.
DNA damage produced by environmental agents is usually removed by DNA repair mechanisms. Individuals who have an inherited inability to repair DNA, as is seen in xeroderma pigmentosa (see Chapter 57) and ataxia telangiectasia, have increased risk of developing a malignancy.
Many Chemicals are Carcinogenic
A wide variety of chemical compounds are carcinogenic (see Table 55-2 and Figure 55–4). It is estimated that perhaps 80% of human cancers are caused by environmental factors, principally chemicals.
TABLE 55–2 Some Chemical Carcinogens
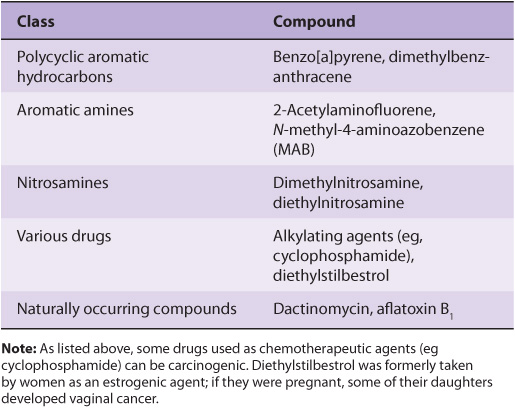
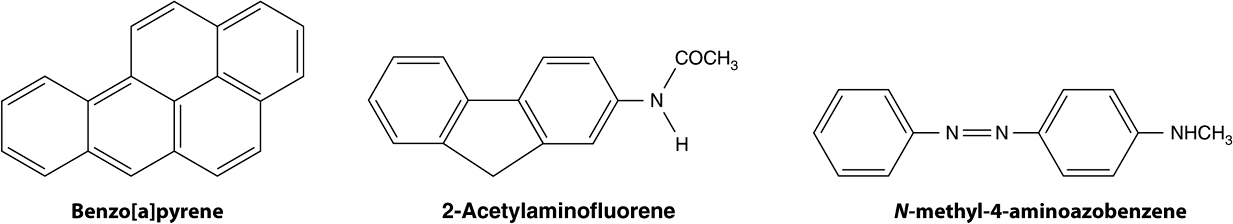
FIGURE 55–4 Structures of three experimentally widely used chemical carcinogens.
Extensive studies have been performed in the field of chemical carcinogenesis. Overall, most chemical carcinogens are thought to interact covalently with DNA, forming a wide variety of adducts. Depending on the extent of damage to DNA and its repair by DNA repair systems (see Chapter 35), a variety of mutations in DNA can result from exposure of an animal or human to chemical carcinogens, some of which contribute to the development of cancer.
Some chemicals interact directly with DNA (eg, methchlorethamine and β-propiolactone), but others (procarcinogens) require conversion by enzyme action to become ultimate carcinogens (Figure 55–5). Most ultimate carcinogens are electrophiles (molecules deficient in electrons) and readily attack nucleophilic (electron-rich) groups in DNA. Conversion of chemicals to ultimate carcinogens is principally due to the actions of various species of cytochrome P450 located in the endoplasmic reticulum (ER) (see Chapter 53). This fact is used in the Ames assay (see below), in which an aliquot of post-mitochondrial supernatant (containing ER) is added to the assay system as a source of cytochrome P450 enzymes.
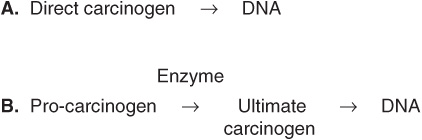
FIGURE 55–5 (A) Direct and (B) indirect carcinogens. Direct carcinogens can interact with DNA without prior enzyme activation. Indirect carcinogens are activated by an enzyme (eg a cytochrome P450 species) to the ultimate carcinogen and then interact with DNA.
Chemical carcinogenesis comprises two stages—initiation and promotion. Initiation is the stage where exposure to a chemical causes irreversible DNA damage and is a necessary initial event for a cell to become cancerous. Promotion comprises the stage at which an initiated cell begins to grow and proliferate. The cumulative effect of these stages is a neoplasm.
Chemical carcinogens can be identified by screening for their mutagenicity. A simple way to do this is by using the Ames assay (Figure 55–6). This relatively simple test, which detects mutations in Salmonella typhimurium caused by chemicals, has proven very valuable for screening purposes. A refinement of the Ames test is to add an aliquot of endoplasmic reticulum (ER) to the assay, to make it possible to indentify procarcino-gens. Very few, if any, compounds that have tested negative in the Ames test have been shown to cause tumors in animals. However, animal testing is required to show unambiguously that a chemical is carcinogenic.
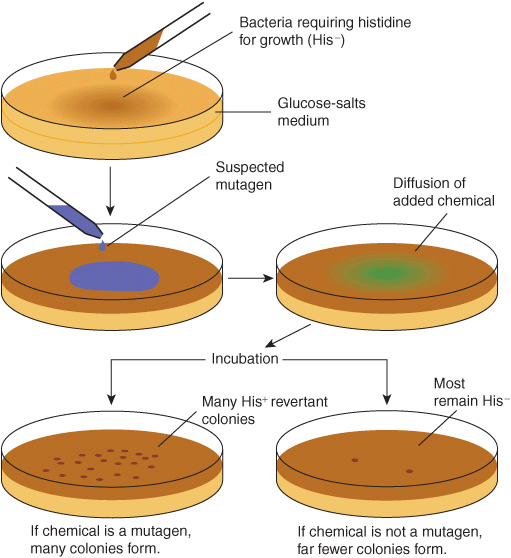
FIGURE 55–6 The Ames assay to screen for mutagens. The chemical tested will increase the frequency of reversion of His–to His+ cells if it is a mutagen and, therefore, a potential carcinogen. A control plate (not shown) contains the liquid in which the suspected mutagen is dissolved. Reproduced, with permission, from Nester EW et al: Microbiology: A Human Perspective. 5th ed. McGraw-Hill, 2007.
It should be noted that compounds that alter epigenetic factors (eg, stilbestrol), thus perhaps leading to cancer, would not test positive in the Ames test, as they are not mutagenic.
Approximately 15% of Human Cancers may be Caused by Viruses
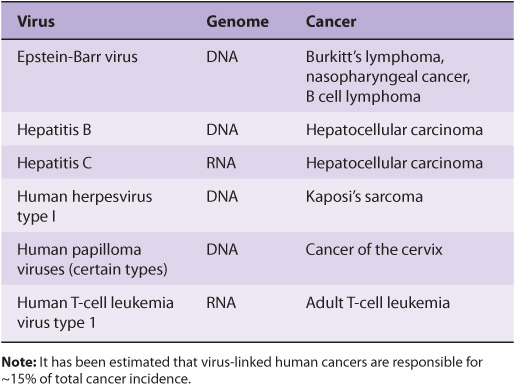
The study of tumor viruses has contributed very significantly to the understanding of cancer. For example, discovery of both oncogenes and tumor suppressor genes (see below) emerged from studies of oncogenic viruses. Both DNA and RNA viruses have been identified as being able to cause cancers in humans (Table 55-3). The details of how each of these viruses causes cancer will not be described here. In general, the genetic material of viruses is incorporated into the genome of the host cell. In the case of RNA viruses, this would occur after reverse transcription of the viral RNA to viral DNA. Such integration of viral DNA (called the provirus) with the host DNA results in various events such as deregulation of the cell cycle, inhibition of apoptosis, and abnormalities of cell signaling pathways. All these events are discussed later in this chapter. The DNA viruses often act by down-regulating the tumor suppressor genes P53 and RB (see below). RNA viruses often carry oncogenes in their genomes; how oncogenes act to cause malignancy is discussed below. It has been estimated that about 15% of human tumors may be caused by viruses.
TABLE 55–3 Some Viruses That Cause or Are Associated With Human Cancers
ONCOGENES AND TUMOR SUPPRESSOR GENES PLAY KEY ROLES IN CAUSING CANCER
Over the past 30 years or so, major advances have been made in understanding how cancer cells develop and grow. Two key findings were the discoveries of oncogenes and tumor suppressor genes. These discoveries pointed to specific mechanisms by which cell growth and division could be disturbed, resulting in abnormal growth. The overall effects of oncogenes and loss of activity of tumor suppressor genes are summarized in Figure 55–7.

FIGURE 55–7 Oncogenes and loss of activity of tumor suppressor genes drive cell growth towards cancer. Oncogenes encode various proteins that can drive the growth of cancer cells. Oncogenes are derived from proto-oncogenes. Tumor suppressor genes encode proteins that normally suppress cell growth, but which are inactivated when altered by mutations. MicroRNA molecules (not shown here) are also affected by mutations, and this can affect their normal regulatory functions. In addition, epigenetic changes (also not shown) affect gene expression, and hence growth of cancer cells.
Oncogenes are Derived from Proto-oncogenes and Encode a Wide Variety of Proteins that Affect Cell Growth and Cell Death
An oncogene can be defined as an altered gene whose product acts in a dominant manner to accelerate cell growth or cell division. It is derived by “activation” of normal cellular proto-oncogenes (which encode growth stimulating proteins). The mechanisms involved in such activation are listed in Table 55-4.
TABLE 55–4 Mechanisms of Activating Oncogenes
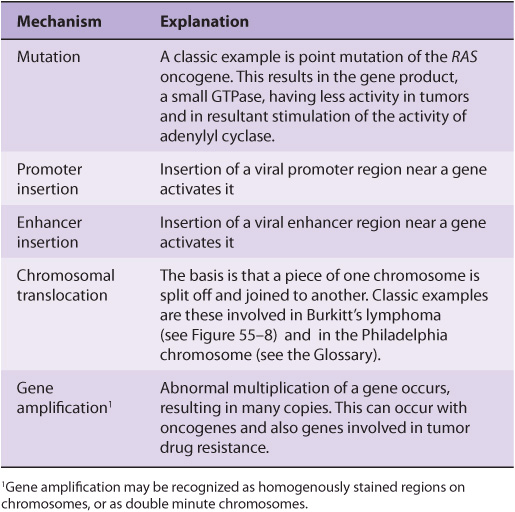
The Table lists an example of a point mutation occurring in the RAS oncogene, which encodes a small GTPase. Loss of the activity of this G protein (see Chapter 42) results in chronic stimulation of the activity of adenylyl cyclase, leading to cell proliferation. Another way an oncogene can be activated is via insertion of a promoter (see Figure 55–8(A)), in which integration of a retroviral provirus (ie, a DNA copy of the RNA genome of a tumor virus such as Rous sarcoma virus, made by reverse transcriptase) activates MYC, a neighboring host gene. Overproduction of the protein encoded by MYC (a transcription factor) stimulates cell proliferation. As the legend to Figure 55–8(A) indicates, a similar type of effect results from enhancer insertion. Chromosomal translocations are found quite frequently in cancer cells, with about a hundred different examples having been documented. The translocation found in cases of Burkitt’s lymphoma is illustrated in Figure 55–8(B). The overall effect of this translocation is also to activate MYC, resulting in cell proliferation. Yet another mechanism of oncogene activation is via gene amplification, which occurs quite commonly in various cancers. In this case, multiple copies of an oncogene are formed resulting in increased production of a growth-promoting protein.
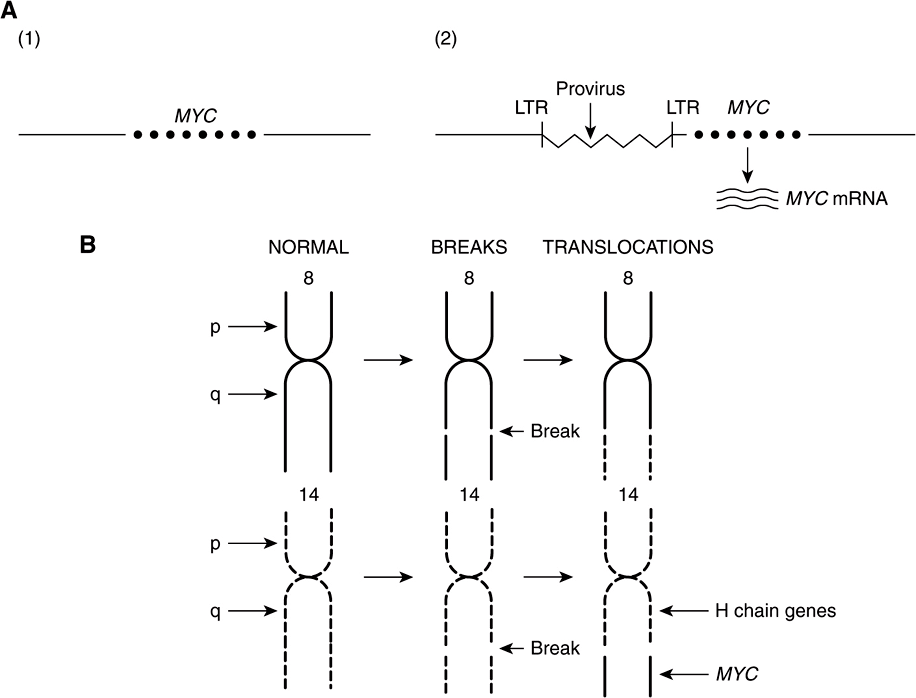
FIGURE 55–8 (A) Schematic representation of how promoter insertion may activate a proto-oncogene. (1). Normal chicken chromosome showing an inactive MYC gene. (2) An avian leukemia virus has integrated in the chromosome in its proviral form (a DNA copy of its RNA genome) adjacent to the MYC gene. Its right-hand long terminal repeat (LTR), containing a strong promoter (see Chapter 36), lies just upstream of the MYC gene and activates that gene, resulting in transcription of MYC mRNA. For simplicity, only one strand of DNA is depicted and other details have been omitted. Enhancer insertion acts similarly, except that the site of integration may be downstream or considerably upstream, and it cannot act as a promoter. Instead, a specific proviral sequence acts as an enhancer element (see Chapter 36), leading to activation of the MYC gene and its transcription. (B) Schematic representation of the reciprocal translocation involved in Burkitt’s lymphoma. The chromosomes involved are 8 and 14. A segment from the end of the q arm of chromosome 8 breaks off and moves to chromosome 14. The reverse process moves a small segment from the q arm of chromosome 14 to chromosome 8. The MYC gene is contained in the small piece of chromosome 8 that was transferred to chromosome 14; it is thus placed next to genes transcribing the heavy chains of immunoglobulin molecules, and itself becomes activated. Many other translocations have been identified, with perhaps the best known being that involved in formation of the Philadelphia chromosome (see the Glossary).
Once oncogenes are activated, how do their protein products act to promote development of cancer? Figure 55–9 shows certain of the ways in which they operate. Some affect cell signaling pathways (eg, the product of an oncogene may act as growth factor, a growth factor receptor, a G-protein or as a downstream signaling molecule). Others act to alter transcription or to deregulate the cell cycle. Yet others can affect cell-cell interactions or the process of apoptosis. These mechanisms help to explain many of the major features of cancer cells shown in Figure 55–1, such as their limitless replicative potential, their signaling defects, their ability to invade and spread, and their evasion of apoptosis.
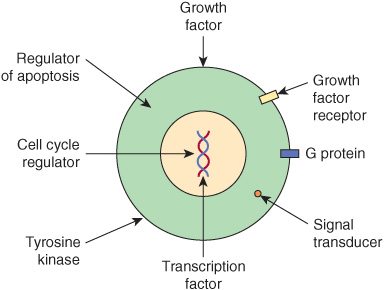
FIGURE 55–9 Some ways in which proteins encoded by oncogenes work. The Figure shows examples of various proteins encoded by oncogenes. The proteins are listed below with the corresponding oncogene given in parentheses along with its OMIM number. A growth factor, fibroblast growth factor 3 (INT2,164950); a growth factor receptor, epidermal growth factor receptor [EGFR] (HER1, 131550); a G protein (H-RAS-1, 190020); a signal transducer (BRAF, 164757); a transcription factor (MYC,190080); a tyrosine kinase and involved in cell-cell adhesion (SRC, 190090); a cell cycle regulator (PRAD, 168461); a regulator of apoptosis (BCL2, 151430).
Certain tumor viruses (eg, retroviruses) contain oncogenes. It was the study of such tumor viruses (eg, Rous sarcoma virus [RSV], a retrovirus) that first revealed the presence of oncogenes. Further study showed that viral oncogenes were derived from cellular proto-oncogenes that the tumor viruses had picked up during their passage through host cells.
Tumor Suppressor Genes Act to Inhibit Cell Growth and Cell Division
A tumor suppressor gene produces a protein product that normally suppresses cell growth or cell division. When such a gene is altered by mutation, the inhibitory effect of its product is lost or diminished, leading to increased cell growth or cell division. As first suggested by AG Knudson, based on studies of the inheritance of retinoblastomas, both copies of a tumor suppressor gene must be affected for it to lose its inhibitory effects on growth.
A useful distinction has been made between gatekeeper and caretaker functions of tumor suppressor genes. The former control cell proliferation, and include mainly genes that act to regulate the cell cycle and apoptosis, The latter are concerned with preserving the integrity of the genome, and include genes whose products are involved in recognizing and correcting DNA damage and maintaining chromosomal integrity during cell division.
Many oncogenes and tumor suppressor genes have now been identified. Only a few are mentioned here. Some differences between oncogenes and tumor suppressor genes are listed in Table 55-5.
TABLE 55–5 Some Differences Between Oncogenes and Tumor Suppressor Genes
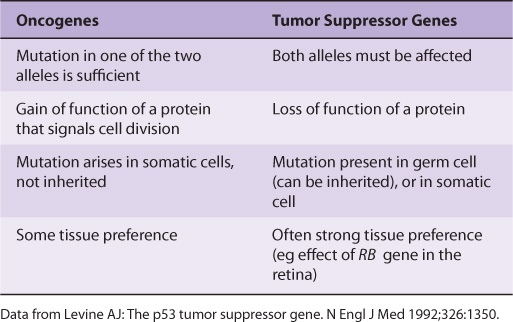
Table 55-6 lists some of the properties of two of the most studied oncogenes (MYC and RAS), and two of the most studied tumor suppressor genes (P53 and RB).
TABLE 55–6 Some Properties of a Few Important Oncogenes and Tumor Suppressor Genes


Studies of the Development of Colorectal Cancers Have Illuminated the Involvements of Specific Oncogenes and Tumor Suppressor Genes
Many types of tumors have been analyzed for genetic changes. One of the most informative areas in this respect has been analyses of the development of colorectal cancers by Vogelstein and colleagues. Their work, and that of others, has shown the involvement of various oncogenes and tumor suppressor genes in human cancer. (Case 4 in Chapter 57 describes the history of a patient with colorectal cancer). These workers analyzed various oncogenes, tumor suppressor genes, and certain other relevant genes in samples of normal colonic epithelium, of dysplastic epithelium (a preneoplastic condition, characterized by abnormal development of epithelium), of various stages of adenomatous polyps, and of adenocarcinomas. Some of their major findings are summarized in Figure 55–10. It can be seen that certain genes were found to be mutated at relatively specific stages of the total sequence shown. Functions of the various genes identified are listed in Table 55-7. The overall sequence of changes can vary somewhat from that shown, and other genes may also be involved. Similar studies have been performed on a number of other human tumors revealing somewhat different patterns of activation of oncogenes and mutations of tumor suppressor genes. Further mutations in these and other genes are involved in tumor progression, a phenomenon whereby clones of tumor cells become selected for fast growth rate and ability to spread. Thus, a relatively large tumor may contain a variety of cells with different genotypes, making successful treatment more difficult.
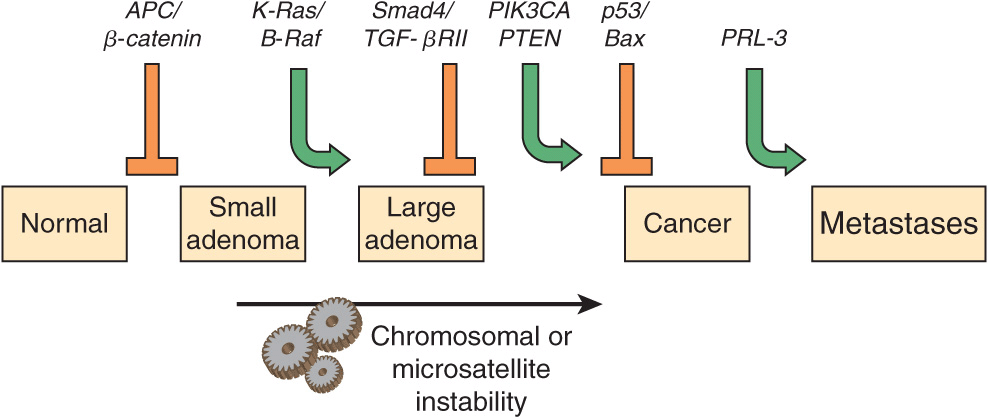
FIGURE 55–10 Multistep genetic changes associated with the development of colorectal cancers. Mutations in the APC gene initiate the formation of adenomas. One sequence of mutations in an oncogene and in various tumor suppressor genes that can result in further progression to large adenomas and cancer is indicated. Patients with familial adenomatous polyposis (OMIM 175100) inherit mutations in the APC gene and develop numerous dysplastic aberrant crypt foci (ACF), some of which progress as they acquire the other mutations indicated in the Figure. The tumors from patients with hereditary nonpolyposis colon cancer (OMIM 120435) go through a similar though not identical series of mutations; mutations in the mismatch repair system (see Chapter 35) speed up this process. K-RAS is an oncogene, and the other specific genes indicated are tumor suppressor genes. The chromosomal locations of the various genes shown here are known. The sequence of events shown here is not invariable in the development of all colorectal cancers. A variety of other genetic alterations have been described in a small fraction of advanced colorectal cancers. These may be responsible for the heterogeneity of biological and clinical properties observed among different cases. Instability of chromosomes and microsatellites (see Chapter 35) occurs in many tumors, and likely involves mutations in a considerable number of genes. (Reproduced, with permission, from Bunz F, Kinzler KW, Vogelstein B: Colorectal Tumors, Fig. 48-2, The Online Metabolic & Molecular Bases of Inherited Disease, www.ommbid.com)
TABLE 55–7 Some Genes Associated with Colorectal Carcinogenesis
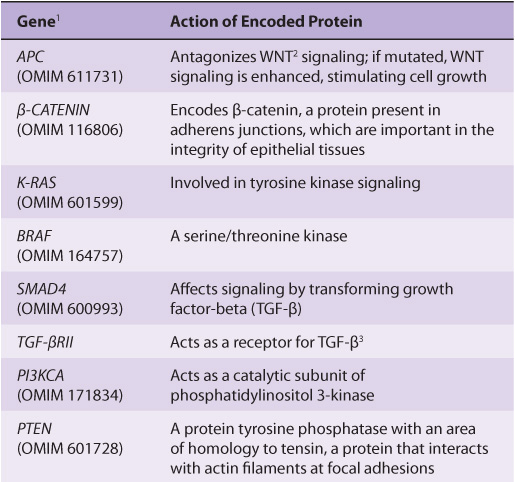
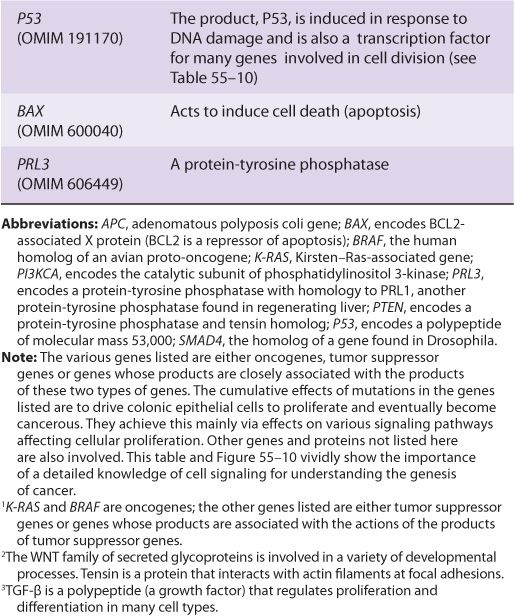
Several other inferences can be made from these results and those from other similar studies. The first of these is that cancer is truly a genetic disease, but in a somewhat different sense from the normal meaning of the phrase, insofar as many of the gene alterations are due to somatic mutations. Secondly, carcinogenesis is, as mentioned above, a multistep process. It is estimated that in most cases a minimum of five to six genes must be mutated for cancer to occur. Thirdly, additional subsequent mutations are thought to confer selective advantages on clones of cells, some of which acquire the ability to metastasize successfully (see below). Fourthly, many of the genes implicated in colorectal carcinogenesis and other types of cancers are involved in cell signaling events, showing the central role that alterations in signaling play in the development of cancer.
GROWTH FACTORS & ABNORMALITIES OF THEIR RECEPTORS AND SIGNALING PATHWAYS PLAY MAJOR ROLES IN CANCER DEVELOPMENT
There Are Many Growth Factors
A large variety of polypeptide growth factors that work on human tissues and cells have been identified. Some are listed in Table 55-8. Here we focus mostly on their relationship with cancer.
TABLE 55–8 Some Polypeptide Growth Factors
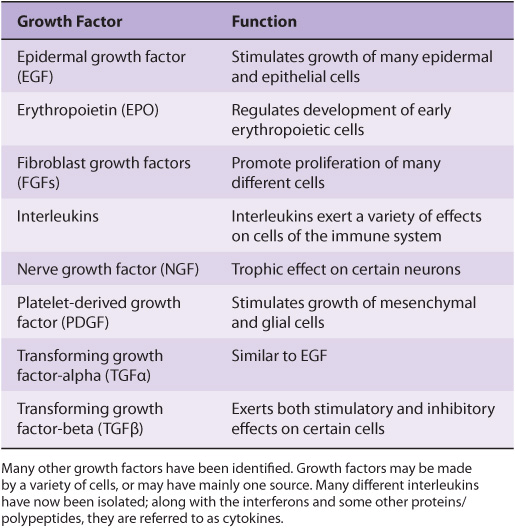
Growth factors can act in an endocrine, paracrine, or autocrine manner and affect a wide variety of cells to produce a mitogenic response. As described earlier (Chapter 52), they play an important role in the differentiation of hematopoietic cells.
Growth inhibitory factors also exist. For example, transforming factor beta (TGF-β) exerts inhibitory effects on the growth of certain cells. Thus, chronic exposure to increased amounts of a growth factor or to decreased amounts of a growth inhibitory factor can alter the balance of cellular growth.
Growth Factors Work Via Specific Receptors and Transmembrane Signaling to Affect the Activities of Specific Genes
Growth factors produce their effects by interacting with specific receptors on cell surfaces, initiating various signaling events (Chapter 42). Many receptors for growth factors have been cloned. They generally have short membrane-spanning segments and external and cytoplasmic domains. A number (eg, those for epidermal growth factor [EGF], insulin and platelet-derived growth factor [PDGF]) have tyrosine kinase activities. The kinase activity, located in the cytoplasmic domains, causes autophosphorylation of the receptor protein and also phosphorylates certain other proteins.
Consideration of how PDGF acts illustrates how one particular growth factor brings about its effects. Interaction of PDGF with its receptor stimulates the activity of phospholipase C. This acts to split phosphatidylinositol bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG) (see Figure 42–6). Increased IP3 stimulates the release of intracellular Ca2+ and DAG increases the activity of protein kinase C (PKC). Hydrolysis of DAG may release arachidonic acid, which can stimulate production of prostaglandins and leukotrienes, each of which has various biologic effects. Exposure of target cells to PDGF can result in rapid (minutes to 1-2 h) activation of certain cellular proto-oncogenes (eg, MYC and FOS), which participate in stimulation of mitosis via effects on the cell cycle (see below). The bottom line is that growth factors interact with specific receptors, which stimulate specific signaling pathways to increase or decrease the activities of various genes that affect cell division.
MANY CANCERS CAN BE PREVENTED BY MODIFYING RISK FACTORS
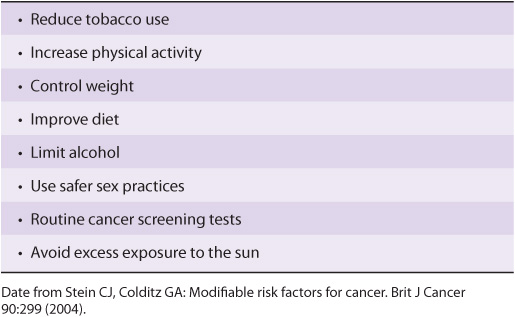
Modifiable risk factors have been linked to a wide variety of cancers. It has been estimated that over half of all cancers in developed countries could be prevented if the measures summarized in Table 55-9 were introduced on a population-wide basis. Smoking is still a major cause of cancer across the globe. It cannot be overemphasized that prevention and early detection of cancer are most critical if the disease is to be beaten.
TABLE 55–9 Measures That Might Prevent Approximately 50% of Cancers if Introduced on a Population-Wide Basis
ABNORMALITIES OF THE CELL CYCLE ARE UBIQUITOUS IN CANCER CELLS
Knowledge of the cell cycle
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


