Molecular Defects More than 90% of patients clinically classified as having MFS by the “Ghent criteria” have a mutation in the gene for fibrillin-1 (FBN1). Mutations in the same gene are found in a few patients who do not meet the Ghent criteria. Also, a few MFS patients without mutations in the FBN1 gene have mutations in the gene for TGF-β receptor 2 (TGFBR2). In addition, mutations in either TGFBR2 or TGFBR1 are found in the related Loeys-Dietz syndrome, which is characterized by aortic aneurysms, cleft palate, and hypertelorism. Mutations in the FBN2 gene, which is structurally similar to the FBN1 gene, are found in patients with MFS-like syndrome of congenital contractual arachnodactyly.
FBN1 gene mutations are scattered throughout its 65 coding exons. Most are private mutations, but about 10% are recurrent new mutations that are largely located in CpG sequences known to be “hot spots.” Most severe mutations are located in the central codons (24–32). About one-third of the mutations introduce premature termination codons, and about two-thirds are missense mutations that alter calcium-binding domains in the repetitive epidermal growth factor–like domains of the protein. Rarer mutations alter the processing of the protein. As in many genetic diseases, the severity of the phenotype cannot be predicted from the nature of the mutation.
The discovery that syndromes similar to MFS are caused by mutations in TGFBR1 and TGFBR2 refocused attention on structural similarity between fibrillin-1 and TGF-β binding proteins that sequester TGF-β in the extracellular matrix. As a result, some of the manifestations of MFS have been shown to arise from alterations in binding sites that modulate TGF-β bioavailability during development of the skeleton and other tissues. Likewise, TGFBR1 and TGFBR2 mutations in Loeys-Dietz syndrome alter TGF-β signaling. In both MFS and Loeys-Dietz syndrome, the pathogenic mechanisms involve increased TGF-β signaling, which contributes to aneurysm formation.
Diagnosis All patients with a suspected diagnosis of MFS should have a slit-lamp examination and an echocardiogram. Also, homocystinuria should be ruled out by amino acid analysis of plasma (Chap. 434e). The diagnosis of MFS according to the international Ghent standards places emphasis on major criteria that include presence of at least four skeletal abnormalities: ectopia lentis; dilation of the ascending aorta with or without dissection; dural ectasia; and a blood relative who meets the same criteria, with or without a DNA diagnosis. A final diagnosis is based on a balanced assessment of the major criteria together with several minor criteria. The absence of ocular changes suggests the Loeys-Dietz syndrome, and the presence of contractures with some of the signs of OI suggests congenital contractual arachnodactyly.
Diagnostic tests based on gene sequencing or detection of protein defects are available. These results are unlikely to alter the treatment or prognosis but are helpful to inform the patients and families and to rapidly exclude the diagnosis in unaffected family members.
TREATMENT | MARFAN’S SYNDROME |
Propranolol or other β-adrenergic blocking agents are used to lower blood pressure and thereby delay or prevent aortic dilation. Surgical correction of the aorta, aortic valve, and mitral valve has been successful in many patients, but tissues are frequently friable. Patients should be advised that the risks are increased by severe physical exertion, emotional stress, and pregnancy.
The scoliosis tends to be progressive and should be treated by mechanical bracing and physical therapy if >20° or by surgery if it progresses to >45°. Dislocated lenses rarely require surgical removal, but patients should be followed closely for retinal detachment. The finding that MFS pathophysiology involves alterations in TGF-β signaling has raised the possibility of new therapeutic strategies. Attenuation of TGF-β signaling with agents such as angiotensin II receptor blockers (e.g., losartan) was effective in animal studies and has been very promising in small observational studies on MFS patients, significantly reducing progressive aortic enlargement. Based on these results, large randomized clinical trials of angiotensin receptor blockers in MFS are under way.
ELASTIN-RELATED DISEASES
Mutations in the elastin gene (ELN) have been found in patients with supravalvular aortic stenosis and skin that hangs in loose and redundant folds (cutis laxa). As indicated in Table 427-3, patients with several forms of EDS have similar changes in skin that were initially thought to reflect changes in elastin.
EPIDERMOLYSIS BULLOSA (EB)
EB has been defined as the category of heritable disorders involving skin that is specifically characterized by blistering as a result of friction. Using this criterion, it was possible to define subtypes by the ultrastructural layer of skin in which the cleavage and blistering occurred. These functional and anatomical criteria made it possible to establish that most patients with a specific subtype have mutations in genes coding for a structural protein, or a cell adherence protein, expressed in the corresponding layer of skin.
Classification and Incidence The four major types of EB are: (1) EB simplex in which cleavage occurs within the epidermis, (2) junctional EB in which cleavage occurs within the lamina lucida, (3) dystrophic EB in which cleavage occurs within the sublamina densa, and (4) Kindler’s syndrome with a mixed level of cleavage in different layers. Patients are then separated into major and minor subtypes based on clinical features and analysis of mutations.
The incidence of EB in the United States is about 1 in 50,000.
![]() Molecular Defects The distinctive anatomic locations in skin have made it possible to relate the clinical subtypes of EB to mutations for specific components. In EB simplex, mutations are found primarily in the genes for the major keratins of basal epithelial cells (keratins 5 and 14) and the cell adhesion proteins plectin, α6β4 integrin, plakophilin-1, and desmoplakin. Patients with the related syndrome, epidermolytic ichthyosis, have mutations in keratin 1 and keratin 10. In junctional EB, mutations occur in type XVII collagen, a laminin (laminin-332), and α6β4 integrin. In the severe syndrome of dystrophic EB, mutations are found in the gene that codes for type VII collagen, which forms long loops anchoring the epidermis to the dermis. Patients with more complex features of what is classified as Kindler’s syndrome have mutations in kindlin-1, a focal adhesion protein involved in integrin activation.
Molecular Defects The distinctive anatomic locations in skin have made it possible to relate the clinical subtypes of EB to mutations for specific components. In EB simplex, mutations are found primarily in the genes for the major keratins of basal epithelial cells (keratins 5 and 14) and the cell adhesion proteins plectin, α6β4 integrin, plakophilin-1, and desmoplakin. Patients with the related syndrome, epidermolytic ichthyosis, have mutations in keratin 1 and keratin 10. In junctional EB, mutations occur in type XVII collagen, a laminin (laminin-332), and α6β4 integrin. In the severe syndrome of dystrophic EB, mutations are found in the gene that codes for type VII collagen, which forms long loops anchoring the epidermis to the dermis. Patients with more complex features of what is classified as Kindler’s syndrome have mutations in kindlin-1, a focal adhesion protein involved in integrin activation.
Diagnosis and Treatment The diagnosis is based on skin that readily breaks and forms blisters from minor trauma. EB simplex is generally milder than junctional EB or dystrophic EB. Dystrophic EB variants usually have large and prominent scars. Precise classification within subtypes usually requires immunofluorescent mapping. DNA diagnostic tests have been developed as research tools but are not widely available. The treatment is symptomatic. Novel therapeutic approaches such as gene therapy, protein replacement therapy, and cell therapy are being explored.
ALPORT’S SYNDROME (AS)
AS is an inherited disorder characterized by hematuria and several associated features. It was not initially considered as a disorder of connective tissue. However, the search for mutations in the genes coding for the collagen found that most patients had mutations in collagen found in basement membranes (type IV). Four forms of AS are now recognized: (1) classic AS, which is inherited as an X-linked disorder with hematuria, sensorineural deafness, and conical deformation of the anterior surface of the lens (lenticonus); (2) an X-linked form associated with diffuse leiomyomatosis; (3) an autosomal recessive form; and (4) an autosomal dominant form. Both autosomal recessive and dominant forms can cause renal disease without deafness or lenticonus.
Incidence The incidence of AS is about 1 in 10,000 births in the general population and as high as 1 in 5000 in some ethnic groups. About 80% of AS patients have the classical X-linked variant.
![]() Molecular Defects Most patients have mutations in four of the six genes for the chains of type IV collagen (COL4A3, COL4A4, COL4A5, and COL4A6). The genes for the proteins are arranged in tandem pairs on different chromosomes in an unusual head-to-head orientation and with overlapping promoters; i.e., the COL4A1 and COL4A2 genes are head-to-head on chromosome 13q34, the COL4A3 and COL4A4 genes are on chromosome 2q35–37, and the COL4A5 and COL4A6 genes are on chromosome Xq22. The X-linked variants are caused by either mutations in the COL4A5 gene or by partial deletions of both of the adjacent COL4A4 and COL4A5 genes. The autosomal recessive variants are caused by mutations in either the COL4A3 or COL4A4 gene. The mutations responsible for the autosomal dominant variants are still unknown, but they have been mapped to the same locus as the COL4A3 and COL4A4 genes.
Molecular Defects Most patients have mutations in four of the six genes for the chains of type IV collagen (COL4A3, COL4A4, COL4A5, and COL4A6). The genes for the proteins are arranged in tandem pairs on different chromosomes in an unusual head-to-head orientation and with overlapping promoters; i.e., the COL4A1 and COL4A2 genes are head-to-head on chromosome 13q34, the COL4A3 and COL4A4 genes are on chromosome 2q35–37, and the COL4A5 and COL4A6 genes are on chromosome Xq22. The X-linked variants are caused by either mutations in the COL4A5 gene or by partial deletions of both of the adjacent COL4A4 and COL4A5 genes. The autosomal recessive variants are caused by mutations in either the COL4A3 or COL4A4 gene. The mutations responsible for the autosomal dominant variants are still unknown, but they have been mapped to the same locus as the COL4A3 and COL4A4 genes.
Diagnosis The diagnosis of classic AS is based on X-linked inheritance of hematuria, sensorineural deafness, and lenticonus. The lenticonus together with hematuria is pathognomonic of classic AS. The sensorineural deafness is primarily in the high-tone range. It can frequently be detected only by an audiogram and is usually not progressive. Because of the X-linked transmission, women are generally underdiagnosed and are usually less severely affected than men. The hematuria usually progresses to nephritis and may cause renal failure in late adolescence in affected males and at older ages in some women. Renal transplantation is usually successful.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Helena Kuivaniemi, Gerard Tromp, Leena Ala-Kokko, and Malwina Czarny-Ratajcak to this chapter in previous editions of Harrison’s. The authors also wish to thank David Sillence for his expert advice on the classifications of types of OI.
428 | Hemochromatosis |
DEFINITION
Hemochromatosis is a common inherited disorder of iron metabolism in which dysregulation of intestinal iron absorption results in deposition of excessive amounts of iron in parenchymal cells with eventual tissue damage and impaired function in a wide range of organs. The iron-storage pigment in tissues is called hemosiderin because it was believed to be derived from the blood. The term hemosiderosis is used to describe the presence of stainable iron in tissues, but tissue iron must be quantified to assess body-iron status accurately (see below and Chap. 126). Hemochromatosis refers to a group of genetic diseases that predispose to iron overload, potentially leading to fibrosis and organ failure. Cirrhosis of the liver, diabetes mellitus, arthritis, cardiomyopathy, and hypogonadotropic hypogonadism are the major clinical manifestations.
Although there is debate about definitions, the following terminology is widely accepted.
1. Hereditary hemochromatosis is most often caused by a mutant gene, termed HFE, which is tightly linked to the HLA-A locus on chromosome 6p (see “Genetic Basis,” below). Persons who are homozygous for the mutation are at increased risk of iron overload and account for 80–90% of clinical hereditary hemochromatosis in persons of northern European descent. In such subjects, the presence of hepatic fibrosis, cirrhosis, arthropathy, or hepatocellular carcinoma constitutes iron overload–related disease. Rarer forms of non-HFE hemochromatosis are caused by mutations in other genes involved in iron metabolism (Table 428-1). The disease can be recognized during its early stages when iron overload and organ damage are minimal. At this stage, the disease is best referred to as early hemochromatosis or precirrhotic hemochromatosis.
CLASSIFICATION OF IRON OVERLOAD STATES |

2. Secondary iron overload occurs as a result of an iron-loading anemia, such as thalassemia or sideroblastic anemia, in which erythropoiesis is increased but ineffective. In the acquired iron-loading disorders, massive iron deposits in parenchymal tissues can lead to the same clinical and pathologic features as in hemochromatosis.
PREVALENCE
HFE-associated hemochromatosis mutations are among the most common inherited disease alleles, although the prevalence varies in different ethnic groups. It is most common in populations of northern European extraction in whom approximately 1 in 10 persons are heterozygous carriers and 0.3–0.5% are homozygotes. However, expression of the disease is variable and modified by several factors, especially alcohol consumption and dietary iron intake, blood loss associated with menstruation and pregnancy, and blood donation. Recent population studies indicate that approximately 30% of homozygous men develop iron overload–related disease and about 6% develop hepatic cirrhosis; for women, the figure is closer to 1%. Presumably there are as yet unidentified modifying genes responsible for expression and there is some early evidence to support this. Nearly 70% of untreated patients develop the first symptoms between ages 40 and 60. The disease is rarely evident before age 20, although with family screening (see “Screening for Hemochromatosis,” below) and periodic health examinations, asymptomatic subjects with iron overload can be identified, including young menstruating women.
In contrast to HFE-associated hemochromatosis, the non-HFE-associated forms of hemochromatosis (Table 428-1) are rare, but they affect all races and young people (juvenile hemochromatosis).
GENETIC BASIS
![]() A homozygous G to A mutation in the HFE gene resulting in a cysteine to tyrosine substitution at position 282 (C282Y) is the most common mutation. It is identified in 85–90% of patients with hereditary hemochromatosis in populations of northern European descent but is found in only 60% of cases from Mediterranean populations (e.g., southern Italy). A second, relatively common HFE mutation (H63D) results in a substitution of histidine to aspartic acid at codon 63. Homozygosity for H63D is not associated with clinically significant iron overload. Some compound heterozygotes (e.g., one copy each of C282Y and H63D) have mild to moderately increased body-iron stores but develop clinical disease only in association with cofactors such as heavy alcohol intake or hepatic steatosis. Thus, HFE-associated hemochromatosis is inherited as an autosomal recessive trait; heterozygotes have no, or minimal, increase in iron stores. However, this slight increase in hepatic iron can act as a cofactor that may modify the expression of other diseases such as porphyria cutanea tarda (PCT) or nonalcoholic steatohepatitis.
A homozygous G to A mutation in the HFE gene resulting in a cysteine to tyrosine substitution at position 282 (C282Y) is the most common mutation. It is identified in 85–90% of patients with hereditary hemochromatosis in populations of northern European descent but is found in only 60% of cases from Mediterranean populations (e.g., southern Italy). A second, relatively common HFE mutation (H63D) results in a substitution of histidine to aspartic acid at codon 63. Homozygosity for H63D is not associated with clinically significant iron overload. Some compound heterozygotes (e.g., one copy each of C282Y and H63D) have mild to moderately increased body-iron stores but develop clinical disease only in association with cofactors such as heavy alcohol intake or hepatic steatosis. Thus, HFE-associated hemochromatosis is inherited as an autosomal recessive trait; heterozygotes have no, or minimal, increase in iron stores. However, this slight increase in hepatic iron can act as a cofactor that may modify the expression of other diseases such as porphyria cutanea tarda (PCT) or nonalcoholic steatohepatitis.
Mutations in other genes involved in iron metabolism are responsible for non-HFE-associated hemochromatosis, including juvenile hemochromatosis, which affects persons in the second and third decades of life (Table 428-1). Mutations in the genes encoding hepcidin, transferrin receptor 2 (TfR2), and hemojuvelin (Fig. 428-1) result in clinicopathologic features that are indistinguishable from HFE-associated hemochromatosis. However, mutations in ferroportin, responsible for the efflux of iron from enterocytes and most other cell types, result in iron loading of reticuloendothelial cells and macrophages as well as parenchymal cells.
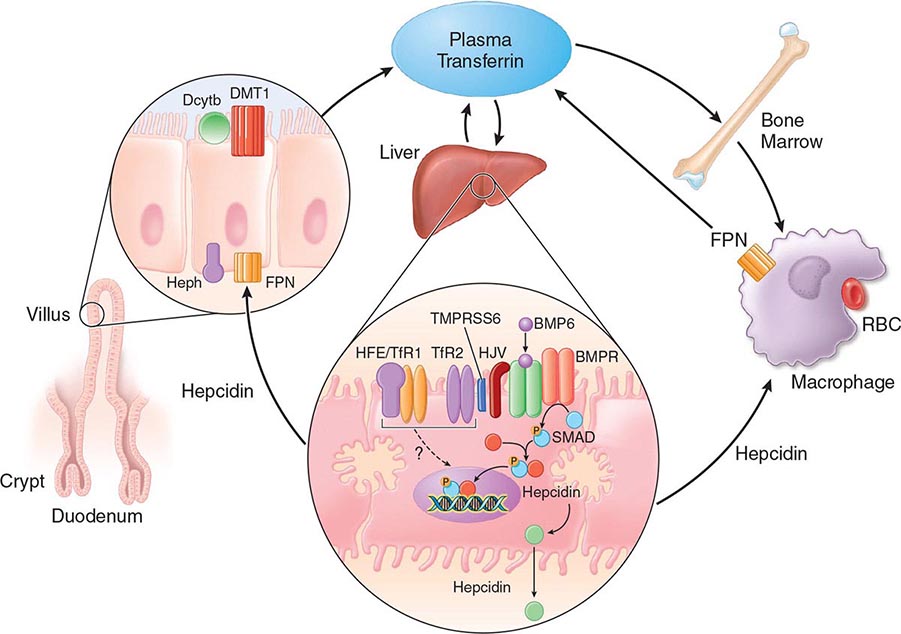
FIGURE 428-1 Pathways of normal iron homeostasis. Dietary inorganic iron traverses the brush border membrane of duodenal enterocytes via the divalent metal-ion transporter 1 (DMT1) after reduction of ferric (Fe3+) iron to the ferrous (Fe2+) state by duodenal cytochrome B (DcytB). Iron then moves from the enterocyte to the circulation via a process requiring the basolateral iron exporter ferroportin (FPN) and the iron oxidase hephaestin (Heph). In the circulation, iron binds to plasma transferrin and is thereby distributed to sites of iron utilization and storage. Much of the diferric transferrin supplies iron to immature erythrocyte cells in the bone marrow for hemoglobin synthesis. At the end of their life, senescent red blood cells (RBCs) are phagocytosed by macrophages, and iron is returned to the circulation after export through ferroportin. The liver-derived peptide hepcidin represses basolateral iron transport in the gut as well as iron released from macrophages and other cells and serves as a central regulator of body-iron traffic. Hepcidin responds to changes in body-iron requirements by signals mediated by diferric transferrin through two mechanisms. One involves HFE and TfR2, whereas the other involves hemojuvelin (HJV) and the bone morphogenetic protein (BMP)/SMAD pathway. TMPRSS6 is a protease that modulates HJV activity. Heme is metabolized by heme oxygenase within the enterocytes, and the released iron then follows the same pathway. Mutations in the genes encoding HFE, TfR2, hemojuvelin, and hepcidin all lead to decreased hepcidin release and increased iron absorption, resulting in hemochromatosis (Table 428-1).
PATHOPHYSIOLOGY
Normally, the body-iron content of 3–4 g is maintained such that intestinal mucosal absorption of iron is equal to iron loss. This amount is approximately 1 mg/d in men and 1.5 mg/d in menstruating women. In hemochromatosis, mucosal absorption is greater than body requirements and amounts to 4 mg/d or more. The progressive accumulation of iron increases plasma iron and saturation of transferrin and results in a progressive increase of plasma ferritin (Fig. 428-2). A liver-derived peptide, hepcidin, represses basolateral iron transport in the intestine and iron release from macrophages and other cells by binding to ferroportin. Hepcidin, in turn, responds to signals in the liver mediated by HFE, TfR2, and hemojuvelin (Fig. 428-1). Thus, hepcidin is a crucial molecule in iron metabolism, linking body stores with intestinal iron absorption.
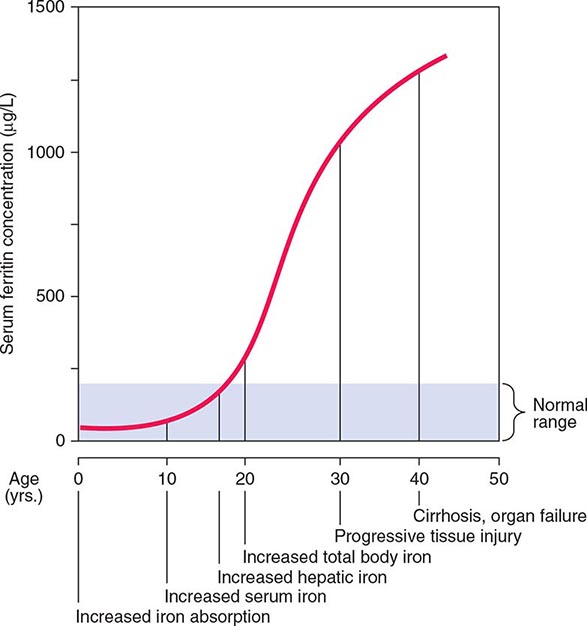
FIGURE 428-2 Sequence of events in genetic hemochromatosis and their correlation with the serum ferritin concentration. Increased iron absorption is present throughout life. Overt, symptomatic disease usually develops between ages 40 and 60, but latent disease can be detected long before this.
The HFE gene encodes a 343-amino-acid protein that is structurally related to MHC class I proteins (HFE). The basic defect in HFE-associated hemochromatosis is a lack of cell surface expression of HFE (due to the C282Y mutation). The normal (wild-type) HFE protein forms a complex with β2-microglobulin and transferrin receptor 1 (TfR1). The C282Y mutation completely abrogates this interaction. As a result, the mutant HFE protein remains trapped intracellularly, reducing TfR1-mediated iron uptake by the intestinal crypt cell. This impaired TfR1-mediated iron uptake leads to upregulation of the divalent metal transporter (DMT1) on the brush border of the villus cells, causing inappropriately increased intestinal iron absorption (Fig. 428-1). In advanced disease, the body may contain 20 g or more of iron that is deposited mainly in parenchymal cells of the liver, pancreas, and heart. Iron may be increased 50- to 100-fold in the liver and pancreas and 5- to 25-fold in the heart. Iron deposition in the pituitary causes hypogonadotropic hypogonadism in both men and women. Tissue injury may result from disruption of iron-laden lysosomes, from lipid peroxidation of subcellular organelles by excess iron, or from stimulation of collagen synthesis by activated stellate cells.
Secondary iron overload with deposition in parenchymal cells occurs in chronic disorders of erythropoiesis, particularly in those due to defects in hemoglobin synthesis or ineffective erythropoiesis such as sideroblastic anemia and thalassemia (Chap. 127). In these disorders, iron absorption is increased. Moreover, these patients require blood transfusions and are frequently treated inappropriately with iron. PCT, a disorder characterized by a defect in porphyrin biosynthesis (Chap. 430), can also be associated with excessive parenchymal iron deposits. The magnitude of the iron load in PCT is usually insufficient to produce tissue damage. However, some patients with PCT also have mutations in the HFE gene, and some have associated hepatitis C virus (HCV) infection. Although the relationship between these disorders remains to be clarified, iron overload accentuates the inherited enzyme deficiency in PCT and should be avoided along with other agents (alcohol, estrogens, haloaromatic compounds) that may exacerbate PCT. Another cause of hepatic parenchymal iron overload is hereditary aceruloplasminemia. In this disorder, impairment of iron mobilization due to deficiency of ceruloplasmin (a ferroxidase) causes iron overload in hepatocytes.
Excessive iron ingestion over many years rarely results in hemochromatosis. An important exception has been reported in South Africa among groups who brew fermented beverages in vessels made of iron. Hemochromatosis has been described in apparently normal persons who have taken medicinal iron over many years, but such individuals probably had genetic disorders.
The common denominator in all patients with hemochromatosis is excessive amounts of iron in parenchymal tissues. Parenteral administration of iron in the form of blood transfusions or iron preparations results predominantly in reticuloendothelial cell iron overload. This appears to lead to less tissue damage than iron loading of parenchymal cells.
In the liver, parenchymal iron is in the form of ferritin and hemosiderin. In the early stages, these deposits are seen in the periportal parenchymal cells, especially within lysosomes in the pericanalicular cytoplasm of the hepatocytes. This stage progresses to perilobular fibrosis and eventually to deposition of iron in bile-duct epithelium, Kupffer cells, and fibrous septa due to activation of stellate cells. In the advanced stage, a macronodular or mixed macro- and micronodular cirrhosis develops. Hepatic fibrosis and cirrhosis correlate significantly with hepatic iron concentration.
At autopsy, the enlarged nodular liver and pancreas are rusty in color. Histologically, iron is increased in many organs, particularly in the liver, heart, and pancreas, and, to a lesser extent, in the endocrine glands. The epidermis of the skin is thin, and melanin is increased in the cells of the basal layer and dermis. Deposits of iron are present around the synovial lining cells of the joints.
CLINICAL MANIFESTATIONS
C282Y homozygotes can be characterized by the stage of progression as follows: (1) a genetic predisposition without abnormalities; (2) iron overload without symptoms; (3) iron overload with symptoms (e.g., arthritis and fatigue); and (4) iron overload with organ damage—in particular, cirrhosis. Thus, many subjects with significant iron overload are asymptomatic. For example, in a study of 672 asymptomatic C282Y homozygous subjects—identified by either family screening or routine health examinations—there was hepatic iron overload (grades 2–4) in 56% and 34.5% of male and female subjects, respectively; hepatic fibrosis (stages 2–4) in 18.4% and 5.4%, respectively; and cirrhosis in 5.6% and 1.9%, respectively.
Initial symptoms are often nonspecific and include lethargy, arthralgia, change in skin color, loss of libido, and features of diabetes mellitus. Hepatomegaly, increased pigmentation, spider angiomas, splenomegaly, arthropathy, ascites, cardiac arrhythmias, congestive heart failure, loss of body hair, testicular atrophy, and jaundice are prominent in advanced disease.
The liver is usually the first organ to be affected, and hepatomegaly is present in more than 95% of symptomatic patients. Hepatic enlargement may exist in the absence of symptoms or of abnormal liver-function tests. Manifestations of portal hypertension and esophageal varices occur less commonly than in cirrhosis from other causes. Hepatocellular carcinoma develops in about 30% of patients with cirrhosis, and it is the most common cause of death in treated patients—hence the importance of early diagnosis and therapy. The incidence increases with age, it is more common in men, and it occurs almost exclusively in cirrhotic patients.
Excessive skin pigmentation is present in patients with advanced disease. The characteristic metallic or slate-gray hue is sometimes referred to as bronzing and results from increased melanin and iron in the dermis. Pigmentation usually is diffuse and generalized, but it may be more pronounced on the face, neck, extensor aspects of the lower forearms, dorsa of the hands, lower legs, and genital regions, as well as in scars.
Diabetes mellitus occurs in about 65% of patients with advanced disease and is more likely to develop in those with a family history of diabetes, suggesting that direct damage to the pancreatic islets by iron deposition occurs in combination with other risk factors. The management is similar to that of other forms of diabetes, although insulin resistance is more common in association with hemochromatosis. Late complications are the same as seen in other causes of diabetes mellitus.
Arthropathy develops in 25–50% of symptomatic patients. It usually occurs after age 50 but may occur as a first manifestation or long after therapy. The joints of the hands, especially the second and third metacarpophalangeal joints, are usually the first joints involved, a feature that helps to distinguish the chondrocalcinosis associated with hemochromatosis from the idiopathic form (Chap. 395). A progressive polyarthritis involving wrists, hips, ankles, and knees may also ensue. Acute brief attacks of synovitis may be associated with deposition of calcium pyrophosphate (chondrocalcinosis or pseudogout), mainly in the knees. Radiologic manifestations include cystic changes of the subchondral bones, loss of articular cartilage with narrowing of the joint space, diffuse demineralization, hypertrophic bone proliferation, and calcification of the synovium. The arthropathy tends to progress despite removal of iron by phlebotomy. Although the relation of these abnormalities to iron metabolism is not known, the fact that similar changes occur in other forms of iron overload suggests that iron is directly involved.
Cardiac involvement is the presenting manifestation in about 15% of symptomatic patients. The most common manifestation is congestive heart failure, which occurs in about 10% of young adults with the disease, especially those with juvenile hemochromatosis. Symptoms of congestive heart failure may develop suddenly, with rapid progression to death if untreated. The heart is diffusely enlarged; this may be misdiagnosed as idiopathic cardiomyopathy if other overt manifestations are absent. Cardiac arrhythmias include premature supraventricular beats, paroxysmal tachyarrhythmias, atrial flutter, atrial fibrillation, and varying degrees of atrioventricular block.
Hypogonadism occurs in both sexes and may antedate other clinical features. Manifestations include loss of libido, impotence, amenorrhea, testicular atrophy, gynecomastia, and sparse body hair. These changes are primarily the result of decreased production of gonadotropins due to impairment of hypothalamic-pituitary function by iron deposition. Adrenal insufficiency, hypothyroidism, and hypoparathyroidism are rare manifestations.
DIAGNOSIS
The association of (1) hepatomegaly, (2) skin pigmentation, (3) diabetes mellitus, (4) heart disease, (5) arthritis, and (6) hypogonadism should suggest the diagnosis. However, as stated above, significant iron overload may exist with none or only some of these manifestations. Therefore, a high index of suspicion is needed to make the diagnosis early. Treatment before permanent organ damage occurs can reverse the iron toxicity and restore life expectancy to normal.
The history should be particularly detailed in regard to disease in other family members; alcohol ingestion; iron intake; and ingestion of large doses of ascorbic acid, which promotes iron absorption (Chap. 96e). Appropriate tests should be performed to exclude iron deposition due to hematologic disease. The presence of liver, pancreatic, cardiac, and joint disease should be confirmed by physical examination, radiography, and standard function tests of these organs.
The degree of increase in total body iron stores can be assessed by (1) measurement of serum iron and the percent saturation of transferrin (or the unsaturated iron-binding capacity), (2) measurement of serum ferritin concentration, (3) liver biopsy with measurement of the iron concentration and calculation of the hepatic iron index (Table 428-2), and (4) magnetic resonance imaging (MRI) of the liver. In addition, a retrospective assessment of body-iron storage is also provided by performing weekly phlebotomy and calculating the amount of iron removed before iron stores are exhausted (1 mL blood = approximately 0.5 mg iron).
REPRESENTATIVE IRON VALUES IN NORMAL SUBJECTS, PATIENTS WITH HEMOCHROMATOSIS, AND PATIENTS WITH ALCOHOLIC LIVER DISEASE |
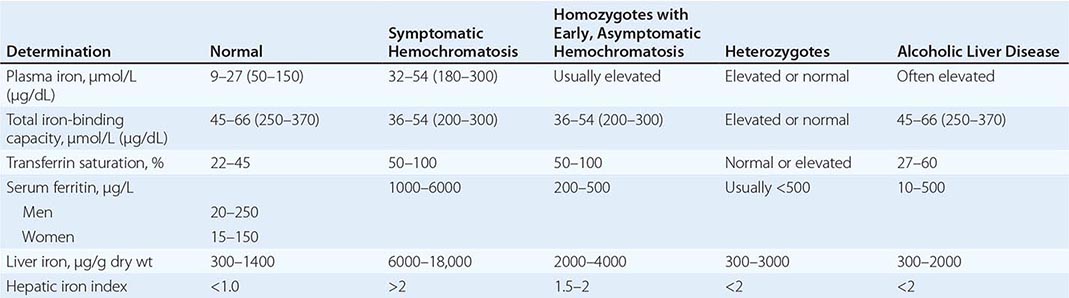
Each of these methods for assessing iron stores has advantages and limitations. The serum iron level and percent saturation of transferrin are elevated early in the course, but their specificity is reduced by significant false-positive and false-negative rates. For example, serum iron concentration may be increased in patients with alcoholic liver disease without iron overload; in this situation, however, the hepatic iron index is usually not increased as in hemochromatosis (Table 428-1). In otherwise healthy persons, a fasting serum transferrin saturation greater than 45% is abnormal and suggests homozygosity for hemochromatosis.
The serum ferritin concentration is usually a good index of body-iron stores, whether decreased or increased. In fact, an increase of 1 μg/L in serum ferritin level reflects an increase of about 5 mg in body stores. In most untreated patients with hemochromatosis, the serum ferritin level is significantly increased (Fig. 428-2 and Table 428-1), and a serum ferritin level >1000 μg/L is the strongest predictor of disease expression among individuals homozygous for the C282Y mutation. However, in patients with inflammation and hepatocellular necrosis, serum ferritin levels may be elevated out of proportion to body-iron stores due to increased release from tissues. Therefore, a repeat determination of serum ferritin should be carried out after acute hepatocellular damage has subsided (e.g., in alcoholic liver disease). Ordinarily, the combined measurements of the percent transferrin saturation and serum ferritin level provide a simple and reliable screening test for hemochromatosis, including the precirrhotic phase of the disease. If either of these tests is abnormal, genetic testing for hemochromatosis should be performed (Fig. 428-3).
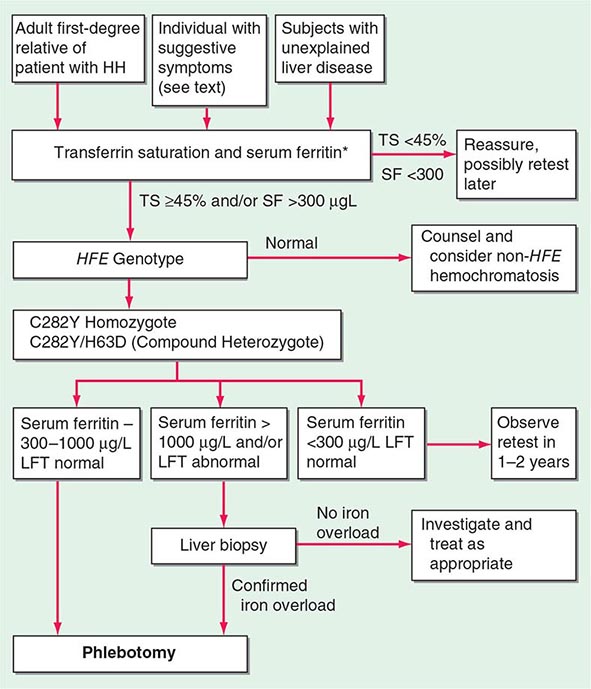
*For convenience both genotype and phenotype (iron tests) can be performed together at a single visit in first-degree relatives.
FIGURE 428-3 Algorithm for screening for HFE-associated hemochromatosis. HH, hereditary hemochromatosis, homozygous subject (C282Y +/+); LFT, liver function tests; SF, serum ferritin concentration; TS, transferrin saturation.
The role of liver biopsy in the diagnosis and management of hemochromatosis has been reassessed as a result of the widespread availability of genetic testing for the C282Y mutation. The absence of severe fibrosis can be accurately predicted in most patients using clinical and biochemical variables. Thus, there is virtually no risk of severe fibrosis in a C282Y homozygous subject with (1) serum ferritin level less than 1000 μg/L, (2) normal serum alanine aminotransferase values, (3) no hepatomegaly, and (4) no excess alcohol intake. However, it should be emphasized that liver biopsy is the only reliable method for establishing or excluding the presence of hepatic cirrhosis, which is the critical factor determining prognosis and the risk of developing hepatocellular carcinoma. Biopsy also permits histochemical estimation of tissue iron and measurement of hepatic iron concentration. Increased density of the liver due to iron deposition can be demonstrated by computed tomography (CT) or MRI, and with improved technology, MRI has become more accurate in determining hepatic iron concentration.
SCREENING FOR HEMOCHROMATOSIS
When the diagnosis of hemochromatosis is established, it is important to counsel and screen other family members (Chap. 84). Asymptomatic and symptomatic family members with the disease usually have an increased saturation of transferrin and an increased serum ferritin concentration. These changes occur even before the iron stores are greatly increased (Fig. 428-2). All adult first-degree relatives of patients with hemochromatosis should be tested for the C282Y and H63D mutations and counseled appropriately (Fig. 428-3). In affected individuals, it is important to confirm or exclude the presence of cirrhosis and begin therapy as early as possible. For children of an identified proband, testing for HFE of the other parent is helpful because if normal, the child is merely an obligate heterozygote and at no risk. Otherwise, for practical purposes, children need not be checked before they are 18 years old.
The role of population screening for hemochromatosis is controversial. Recent studies indicate that it is highly effective for primary care physicians to screen subjects using transferrin saturation and serum ferritin levels. Such screening also detects iron deficiency. Genetic screening of the normal population is feasible but is probably not cost effective.
TREATMENT | HEMOCHROMATOSIS |
The therapy of hemochromatosis involves removal of the excess body iron and supportive treatment of damaged organs. Iron removal is best accomplished by weekly or twice-weekly phlebotomy of 500 mL. Although there is an initial modest decline in the volume of packed red blood cells to about 35 mL/dL, the level stabilizes after several weeks. The plasma transferrin saturation remains increased until the available iron stores are depleted. In contrast, the plasma ferritin concentration falls progressively, reflecting the gradual decrease in body-iron stores. One 500-mL unit of blood contains 200–250 mg of iron, and up to 25 g of iron or more may have to be removed. Therefore, in patients with advanced disease, weekly phlebotomy may be required for 1–2 years, and it should be continued until the serum ferritin level is <50 μg/L. Thereafter, phlebotomies are performed at appropriate intervals to maintain ferritin levels between 50 and 100 μg/L. Usually one phlebotomy every 3 months will suffice.
Chelating agents such as deferoxamine, when given parenterally, remove 10–20 mg of iron per day, which is much less than that mobilized by once-weekly phlebotomy. Phlebotomy is also less expensive, more convenient, and safer for most patients. However, chelating agents are indicated when anemia or hypoproteinemia is severe enough to preclude phlebotomy. Subcutaneous infusion of deferoxamine using a portable pump is the most effective means of its administration.
An effective oral iron chelating agent, deferasirox (Exjade), has recently become available but is still in clinical trials. This agent is effective in thalassemia and secondary iron overload, but its role in primary iron overload has yet to be established.
Alcohol consumption should be severely curtailed or eliminated because it increases the risk of cirrhosis in hereditary hemochromatosis nearly tenfold. Dietary adjustments are unnecessary, although vitamin C and iron supplements should be avoided. The management of hepatic failure, cardiac failure, and diabetes mellitus is similar to conventional therapy for these conditions. Loss of libido and change in secondary sex characteristics are managed with testosterone replacement or gonadotropin therapy (Chap. 411).
End-stage liver disease may be an indication for liver transplantation, although results are improved if the excess iron can be removed beforehand. The available evidence indicates that the fundamental metabolic abnormality in hemochromatosis is reversed by successful liver transplantation.
PROGNOSIS
The principal causes of death are cardiac failure, hepatocellular failure or portal hypertension, and hepatocellular carcinoma.
Life expectancy is improved by removal of the excessive stores of iron and maintenance of these stores at near-normal levels. The 5-year survival rate with therapy increases from 33 to 89%. With repeated phlebotomy, the liver decreases in size, liver function improves, pigmentation of skin decreases, and cardiac failure may be reversed. Diabetes improves in about 40% of patients, but removal of excess iron has little effect on hypogonadism or arthropathy. Hepatic fibrosis may decrease, but established cirrhosis is irreversible. Hepatocellular carcinoma occurs as a late sequela in patients who are cirrhotic at presentation. The apparent increase in its incidence in treated patients is probably related to their increased life span. Hepatocellular carcinoma rarely develops if the disease is treated in the precirrhotic stage. Indeed, the life expectancy of homozygotes treated before the development of cirrhosis is normal.
The importance of family screening and early diagnosis and treatment cannot be overemphasized. Asymptomatic individuals detected by family studies should have phlebotomy therapy if iron stores are moderately to severely increased. Assessment of iron stores at appropriate intervals is also important. With this management approach, most manifestations of the disease can be prevented.
ROLE OF HFE MUTATIONS IN OTHER LIVER DISEASES
![]() There is considerable interest in the role of HFE mutations and hepatic iron in several other liver diseases. Several studies have shown an increased prevalence of HFE mutations in PCT patients. Iron accentuates the inherited enzyme deficiency in PCT and clinical manifestations of PCT. The situation in nonalcoholic steatohepatitis (NASH) is less clear, but some studies have shown an increased prevalence of HFE mutations in NASH patients. The role of phlebotomy therapy, however, is unproven. In chronic HCV infection, HFE mutations are not more common, but some subjects have increased hepatic iron. Before initiating antiviral therapy in these patients, it is reasonable to perform phlebotomy therapy to remove excess iron stores, because this reduces liver enzyme levels.
There is considerable interest in the role of HFE mutations and hepatic iron in several other liver diseases. Several studies have shown an increased prevalence of HFE mutations in PCT patients. Iron accentuates the inherited enzyme deficiency in PCT and clinical manifestations of PCT. The situation in nonalcoholic steatohepatitis (NASH) is less clear, but some studies have shown an increased prevalence of HFE mutations in NASH patients. The role of phlebotomy therapy, however, is unproven. In chronic HCV infection, HFE mutations are not more common, but some subjects have increased hepatic iron. Before initiating antiviral therapy in these patients, it is reasonable to perform phlebotomy therapy to remove excess iron stores, because this reduces liver enzyme levels.
HFE mutations are not increased in frequency in alcoholic liver disease. Hemochromatosis in a heavy drinker can be distinguished from alcoholic liver disease by the presence of the C282Y mutation.
End-stage liver disease may also be associated with iron overload of the degree seen in hemochromatosis. The mechanism is uncertain, although studies have shown that alcohol suppresses hepatic hepcidin secretion. Hemolysis also plays a role. HFE mutations are uncommon.
A recent large population study has suggested that subjects homozygous for C282Y are at increased risk of breast and colorectal cancer.
GLOBAL CONSIDERATIONS
 The HFE mutation is of northern European origin (Celtic or Nordic) with a heterozygous carrier rate of approximately 1 in 10 (1 in 8 in Ireland). Thus, HFE-associated hemochromatosis is quite rare in non-European populations, e.g., Asia. However, non-HFE-associated hemochromatosis resulting from mutations in other genes involved in iron metabolism (Fig. 428-1) is ubiquitous and should be considered when one encounters iron overload.
The HFE mutation is of northern European origin (Celtic or Nordic) with a heterozygous carrier rate of approximately 1 in 10 (1 in 8 in Ireland). Thus, HFE-associated hemochromatosis is quite rare in non-European populations, e.g., Asia. However, non-HFE-associated hemochromatosis resulting from mutations in other genes involved in iron metabolism (Fig. 428-1) is ubiquitous and should be considered when one encounters iron overload.
429 | Wilson’s Disease |
Wilson’s disease is an autosomal recessive disorder caused by mutations in the ATP7B gene, which encodes a membrane-bound, copper-transporting ATPase. Clinical manifestations are caused by copper toxicity and primarily involve the liver and the brain. Because effective treatment is available, it is important to make this diagnosis early.
The frequency of Wilson’s disease in most populations is about 1 in 30,000–40,000, and the frequency of carriers of ATP7B mutations is ∼1%. Siblings of a diagnosed patient have a 1 in 4 risk of Wilson’s disease, whereas children of an affected patient have about a 1 in 200 risk. Because a large number of inactivating mutations have been reported in the ATP7B gene, mutation screening for diagnosis is not routine, although this approach may be practical in the future. DNA haplotype analysis can be used to genotype siblings of an affected patient. A rare multisystem disorder of copper metabolism with features of both Menkes and Wilson’s diseases has been reported. It is termed the MEDNIK syndrome (mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia) and is caused by mutations in the AP1S1 gene, which encodes an adaptor protein necessary for intracellular trafficking of copper pump proteins ATP7A (Menkes disease) and ATP7B (Wilson’s disease).
PATHOGENESIS
ATP7B protein deficiency impairs biliary copper excretion, resulting in positive copper balance, hepatic copper accumulation, and copper toxicity from oxidant damage. Excess hepatic copper is initially bound to metallothionein; liver damage begins as this storage capacity is exceeded, sometimes by 3 years of age. Defective copper incorporation into apoceruloplasmin leads to excess catabolism and low blood levels of ceruloplasmin. Serum copper levels are usually lower than normal because of low blood levels of ceruloplasmin, which normally binds >90% of serum copper. As the disease progresses, nonceruloplasmin serum copper (“free” copper) levels increase, resulting in copper buildup in other parts of the body (e.g., in the brain, with consequent neurologic and psychiatric disease).
CLINICAL PRESENTATION
Hepatic Features Wilson’s disease may present as hepatitis, cirrhosis, or hepatic decompensation. Patients typically present in the mid- to late teenage years in Western countries, although the age of presentation is quite broad and extends into the fifth decade of life. An episode of hepatitis may occur—with elevated serum aminotransferase levels, with or without jaundice—and then spontaneously regress. Hepatitis often recurs, and most of these patients eventually develop cirrhosis. Hepatic decompensation is associated with elevated serum bilirubin, reduced serum albumin and coagulation factors, ascites, peripheral edema, and hepatic encephalopathy. In severe hepatic failure, hemolytic anemia may develop because large amounts of copper derived from hepatocellular necrosis are released into the bloodstream. The association of hemolysis and liver disease makes Wilson’s disease a likely diagnosis.
Neurologic Features The neurologic manifestations of Wilson’s disease typically occur in patients in their early twenties, although the age of onset extends into the sixth decade of life. MRI and CT scans reveal damage in the basal ganglia and occasionally in the pons, medulla, thalamus, cerebellum, and subcortical areas. The three main movement disorders include dystonia, incoordination, and tremor. Dysarthria and dysphagia are common. In some patients, the clinical picture closely resembles that of Parkinson’s disease. Dystonia can involve any part of the body and eventually leads to grotesque positions of the limbs, neck, and trunk. Autonomic disturbances may include orthostatic hypotension and sweating abnormalities as well as bowel, bladder, and sexual dysfunction. Memory loss, migraine-type headaches, and seizures may occur. Patients have difficulty focusing on tasks, but cognition usually is not grossly impaired. Sensory abnormalities and muscular weakness are not features of the disease.
Psychiatric Features Half of patients with neurologic disease have a history of behavioral disturbances with onset in the 5 years before diagnosis. The features are diverse and may include loss of emotional control (temper tantrums, crying bouts), depression, hyperactivity, or loss of sexual inhibition.
Other Manifestations Some female patients have repeated spontaneous abortions, and most become amenorrheic prior to diagnosis. Cholelithiasis and nephrolithiasis occur with increased frequency. Some patients have osteoarthritis, particularly of the knee. Microscopic hematuria is common, and levels of urinary excretion of phosphates, amino acids, glucose, or urates may increase; however, a full-blown Fanconi syndrome is rare. Sunflower cataracts and Kayser-Fleischer rings (copper deposits in the outer rim of the cornea) may be seen. Electrocardiographic and other cardiac abnormalities have been reported but are not common.
DIAGNOSIS
Diagnostic tests for Wilson’s disease are listed in Table 429-1. Serum ceruloplasmin levels should not be used for definitive diagnosis, because they are normal in up to 10% of affected patients and are reduced in 20% of carriers. Kayser-Fleischer rings (Fig. 429-1) can be definitively diagnosed only by an ophthalmologist using a slit lamp. They are present in >99% of patients with neurologic/psychiatric forms of the disease and have been described very rarely in the absence of Wilson’s disease. Kayser-Fleischer rings are present in only ∼30–50% of patients diagnosed in the hepatic or presymptomatic state; thus, the absence of rings does not exclude the diagnosis.
USEFUL TESTS FOR WILSON’S DISEASE |
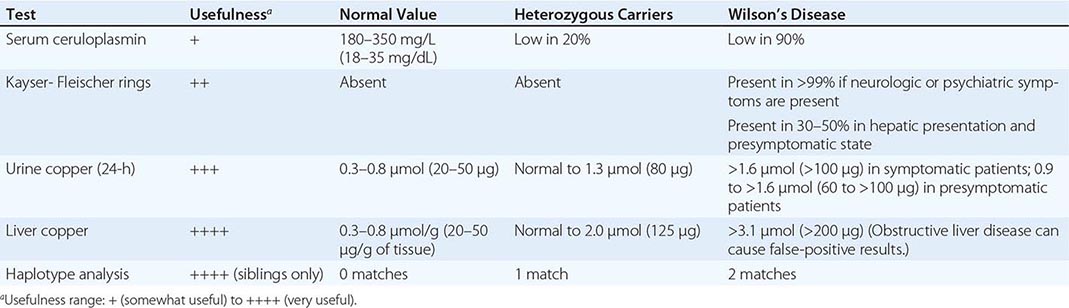
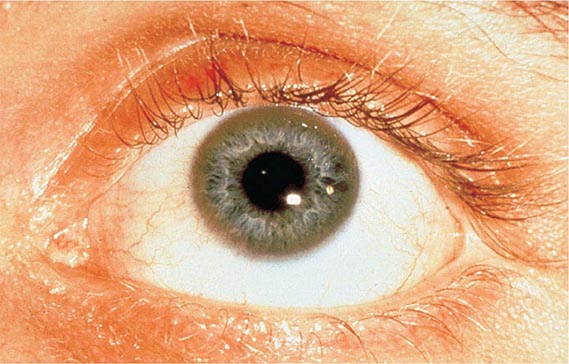
FIGURE 429-1 A Kayser-Fleischer ring. Although in this case, the brownish ring rimming the cornea is clearly visible to the naked eye, confirmation is usually made by slit-lamp examination.
Urine copper measurement is an important diagnostic tool, but urine must be collected carefully to avoid contamination. Symptomatic patients invariably have urine copper levels >1.6 μmol (>100 μg) per 24 h. Heterozygotes have values <1.3 μmol (<80 μg) per 24 h. About half of presymptomatic patients who are ultimately affected have diagnostically elevated urine copper values, but the other half have levels that are in an intermediate range between 0.9 and 1.6 μmol (60–100 μg) per 24 h. Because heterozygotes may have values up to 1.3 μmol (80 μg) per 24 h, patients in this range may require a liver biopsy for definitive diagnosis.
The gold standard for diagnosis remains liver biopsy with quantitative copper assays. Affected patients have values >3.1 μmol/g (>200 μg/g [dry weight] of liver). Copper stains are not reliable. False-positive results can occur with long-standing obstructive liver disease, which can elevate hepatic and urine copper concentrations and rarely causes Kayser-Fleischer rings.
TREATMENT | WILSON’S DISEASE |
Recommended anticopper treatments are listed in Table 429-2. Penicillamine was previously the primary anticopper treatment but now plays only a minor role because of its toxicity and because it often worsens existing neurologic disease if used as initial therapy. If penicillamine is given, it should always be accompanied by pyridoxine (25 mg/d). Trientine is a less toxic chelator and is supplanting penicillamine when a chelator is indicated.
RECOMMENDED ANTICOPPER DRUGS FOR WILSON’S DISEASE |
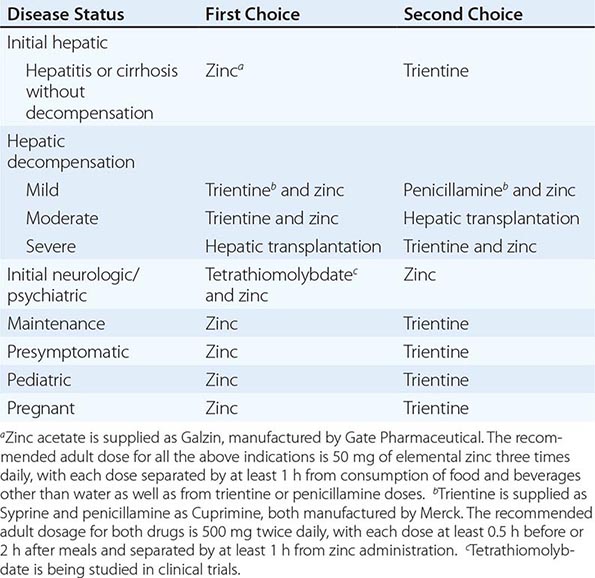
For patients with hepatitis or cirrhosis but without evidence of hepatic decompensation or neurologic/psychiatric symptoms, zinc is the therapy of choice although some experts advocate therapy with trientine. Zinc has proven efficacy in Wilson’s disease and is essentially nontoxic. It produces a negative copper balance by blocking intestinal absorption of copper, and it induces hepatic metallothionein synthesis, thereby sequestering additional toxic copper. All presymptomatic patients should be treated prophylactically because the disease is close to 100% penetrant.
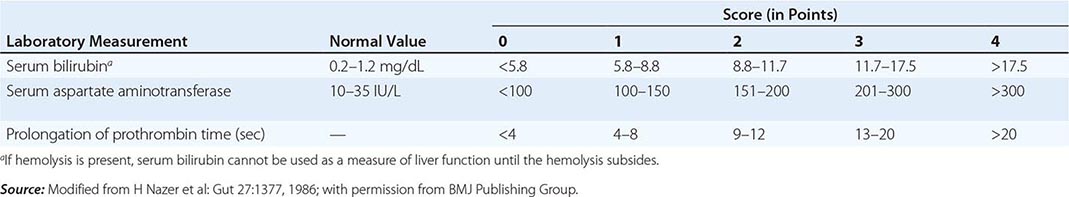
The first step in evaluating patients presenting with hepatic decompensation is to establish disease severity, which can be estimated with the Nazer prognostic index (Table 429-3). Patients with scores <7 can usually be managed with medical therapy. Patients with scores >9 should be considered immediately for liver transplantation. For patients with scores between 7 and 9, clinical judgment is required in deciding whether to recommend transplantation or medical therapy. A combination of trientine and zinc has been used to treat patients with Nazer scores as high as 9, but such patients should be watched carefully for indications of hepatic deterioration, which mandates transplantation.
PROGNOSTIC INDEX OF NAZER |
For initial medical treatment of patients with hepatic decompensation, the recommended regimen is a chelator (preferably trientine) plus zinc (Table 429-2). Zinc should not, however, be ingested simultaneously with trientine, which chelates zinc and forms therapeutically ineffective complexes. Administration of the two drugs should be separated by at least 1 h.
For initial neurologic therapy, tetrathiomolybdate is emerging as the drug of choice because of its rapid control of free copper, preservation of neurologic function, and low toxicity. Penicillamine and trientine should be avoided because both have a high risk of worsening the neurologic condition. Until tetrathiomolybdate is commercially available, zinc therapy is recommended. Although it is relatively slow-acting, zinc itself does not exacerbate neurologic abnormalities. Although hepatic transplantation may alleviate neurologic symptoms, it does so only by copper removal, which can be done more safely and inexpensively with anticopper drugs. Pregnant patients should be treated with zinc or trientine throughout pregnancy but without tight copper control because copper deficiency can be teratogenic.
Anticopper therapy must be lifelong. With treatment, liver function usually recovers after about a year although residual liver damage is usually present. Neurologic and psychiatric symptoms usually improve after 6–24 months of treatment.
MONITORING ANTICOPPER THERAPY
When trientine or penicillamine is first used, it is necessary to monitor for drug toxicity, particularly bone marrow suppression and proteinuria. Complete blood counts, standard biochemical profiles, and a urinalysis should be performed at weekly intervals for 1 month, then at twice-weekly intervals for 2 or 3 months, then at monthly intervals for 3 or 4 months, and at 4- to 6-month intervals thereafter.
The anticopper effects of trientine and penicillamine can be monitored by following 24-h “free” serum copper levels. Changes in urine copper levels are more difficult to interpret because excretion reflects the effect of the drug as well as body loading with copper. Free serum copper is calculated by subtracting the ceruloplasmin copper from the total serum copper. Each 10 mg/L (1 mg/dL) of ceruloplasmin contributes 0.5 μmol/L (3 μg/dL) of serum copper. The normal serum free copper value is 1.6–2.4 μmol/L (10–15 μg/dL); the level is often as high as 7.9 μmol/L (50 μg/dL) in untreated Wilson’s disease. With treatment, the serum free copper should be <3.9 μmol/L (<25 μg/dL).
Zinc treatment does not require monitoring of blood or urine for toxicity. Its only significant side effect is gastric burning or nausea in ∼10% of patients, usually with the first morning dose. This effect can be mitigated if the first dose is taken an hour after breakfast or if zinc is taken with a small amount of protein. Because zinc mainly affects stool copper, 24-h urine copper can be used to reflect body loading. The typical value in untreated symptomatic patients is >3.1 μmol (>200 μg) per 24 h. This level should decrease during the first 1–2 years of therapy to <2.0 μmol (<125 μg) per 24 h. A normal value (0.3–0.8 μmol [20–50 μg]) is rarely reached during the first decade of therapy and should raise concern about overtreatment (copper deficiency), the first sign of which is anemia and/or leukopenia.
GLOBAL CONSIDERATIONS
 The age of onset of clinical disease may be considerably younger in India and the Far East; in these regions, onset often occurs in children at only 5 or 6 years of age. The incidence of the disease may be increased in certain populations as a result of founder effects. For example, in Sardinia, the incidence may be 1 in 3000. In countries where penicillamine, trientine, and zinc acetate (as Galzin) are not available or are unaffordable, zinc salts such as gluconate or sulfate provide an alternative treatment option.
The age of onset of clinical disease may be considerably younger in India and the Far East; in these regions, onset often occurs in children at only 5 or 6 years of age. The incidence of the disease may be increased in certain populations as a result of founder effects. For example, in Sardinia, the incidence may be 1 in 3000. In countries where penicillamine, trientine, and zinc acetate (as Galzin) are not available or are unaffordable, zinc salts such as gluconate or sulfate provide an alternative treatment option.
430 | The Porphyrias |
The porphyrias are metabolic disorders, each resulting from the deficiency of a specific enzyme in the heme biosynthetic pathway (Fig. 430-1 and Table 430-1). These enzyme deficiencies are inherited as autosomal dominant, autosomal recessive, or X-linked traits, with the exception of porphyria cutanea tarda (PCT), which usually is sporadic (Table 430-1). The porphyrias are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of their respective porphyrin precursors or porphyrins (Tables 430-1 and 430-2), although some have overlapping features. For example, PCT, the most common porphyria, is hepatic and presents with blistering cutaneous photosensitivity, which is typically characteristic of the erythropoietic porphyrias. The major manifestations of the acute hepatic porphyrias are neurologic, including neuropathic abdominal pain, peripheral motor neuropathy, and mental disturbances, with attacks often precipitated by dieting, certain drugs, and hormonal changes. While hepatic porphyrias are symptomatic primarily in adults, rare homozygous variants of the autosomal dominant hepatic porphyrias usually manifest clinically prior to puberty.
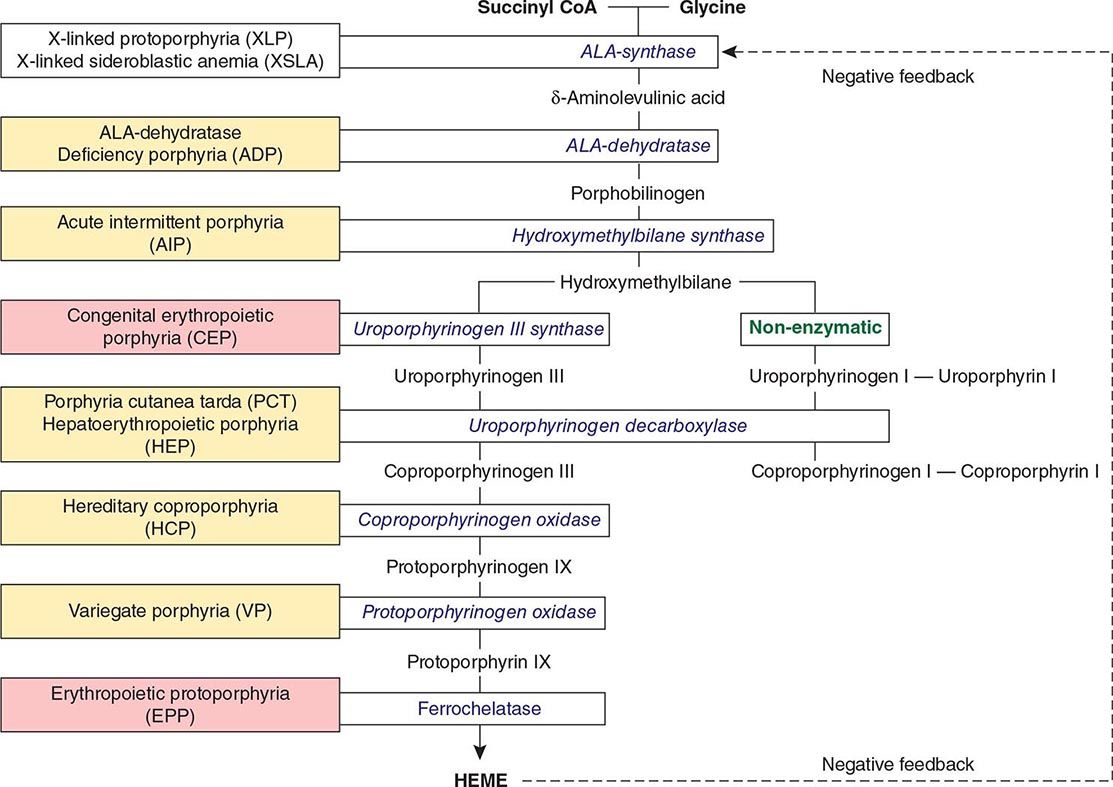
FIGURE 430-1 The human heme biosynthetic pathway indicating in linked boxes the enzyme that, when deficient, causes the respective porphyria. Hepatic porphyrias are shown in yellow boxes and erythropoietic porphyrias in pink boxes.
HUMAN PORPHYRIAS: MAJOR CLINICAL AND LABORATORY FEATURES |
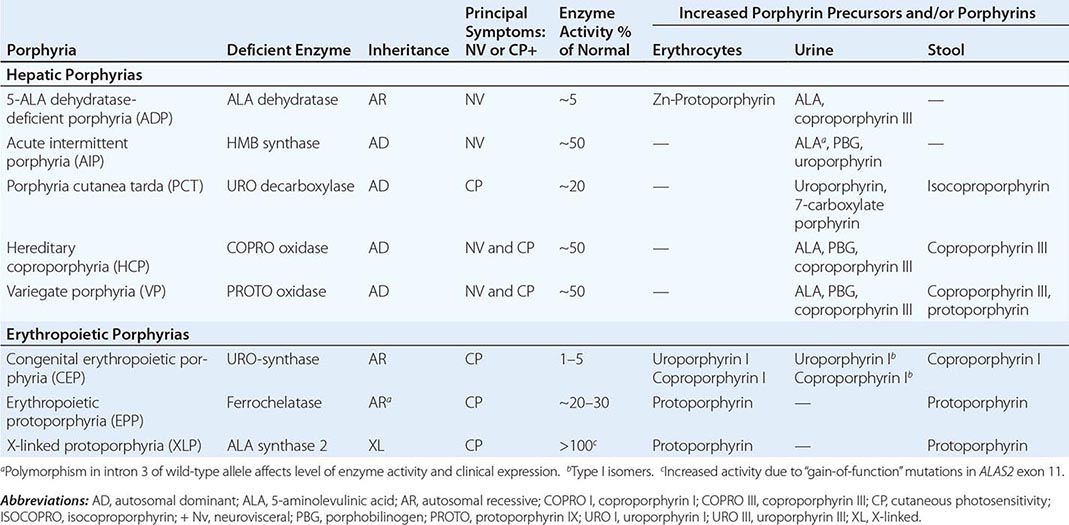
HUMAN HEME BIOSYNTHETIC ENZYMES AND GENES |
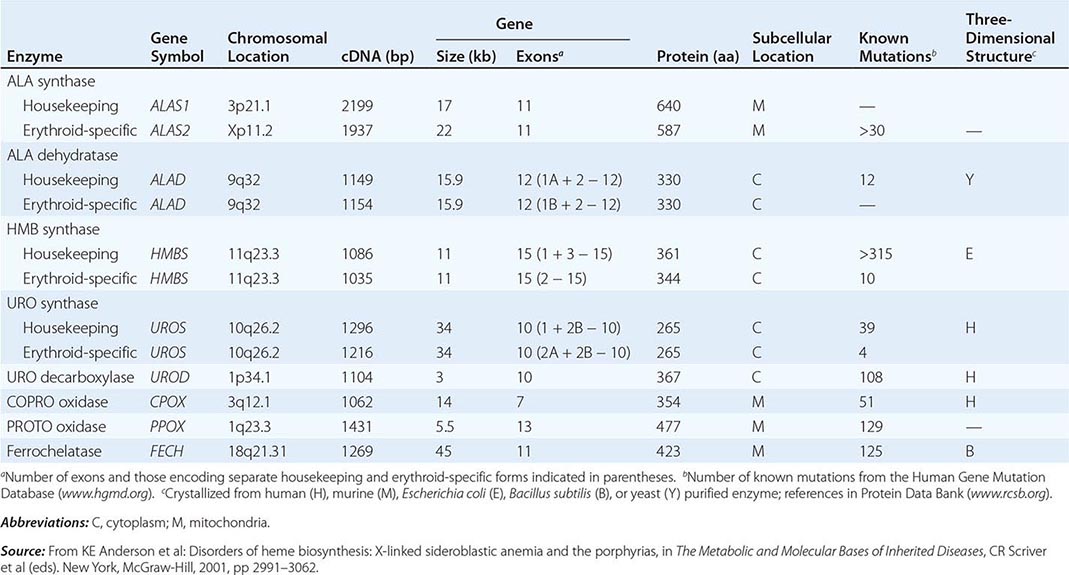
In contrast, the erythropoietic porphyrias usually present at birth or in early childhood with cutaneous photosensitivity, or in the case of congenital erythropoietic porphyria (CEP), even in utero as nonimmune hydrops fetalis. Cutaneous sensitivity to sunlight results from excitation of excess porphyrins in the skin by long-wave ultraviolet light, leading to cell damage, scarring, and disfigurement. Thus, the porphyrias are metabolic disorders in which environmental, physiologic, and genetic factors interact to cause disease.
Because many symptoms of the porphyrias are nonspecific, diagnosis is often delayed. Laboratory measurement of porphyrin precursors (5′-aminolevulinic acid [ALA] and porphobilinogen [PBG]) or porphyrins in urine, plasma, erythrocytes, or feces is required to confirm or exclude the various types of porphyria (see below). However, a definite diagnosis requires demonstration of the specific gene defect (Table 430-3). The genes encoding all the heme biosynthetic enzymes have been characterized, permitting identification of the mutations causing each porphyria (Table 430-2). Molecular genetic analyses now make it possible to provide precise heterozygote or homozygote identification and prenatal diagnoses in families with known mutations.
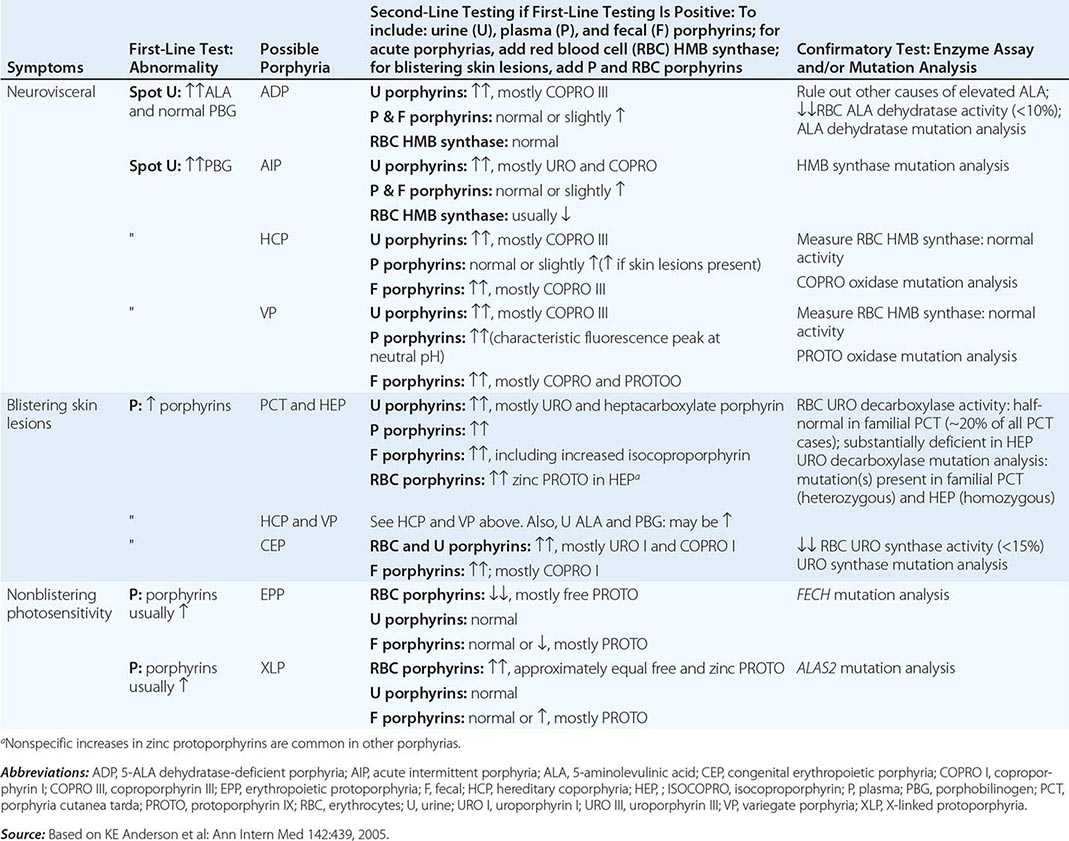
DIAGNOSIS OF ACUTE AND CUTANEOUS PORPHYRIAS |
In addition to recent reviews of the porphyrias, informative and up-to-date websites are sponsored by the American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Initiative (www.porphyria-europe.org). An extensive list of unsafe and safe drugs for individuals with acute porphyrias is provided at the Drug Database for Acute Porphyrias (www.drugs-porphyria.com).
GLOBAL CONSIDERATIONS
 The porphyrias are panethnic metabolic diseases that affect individuals around the globe. The acute hepatic porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—are autosomal dominant disorders. The frequency of AIP, the most common acute hepatic porphyria, is ~1 in 20,000 among Caucasian individuals of Western European ancestry, and it is particularly frequent in Scandinavians, with a frequency of ~1 in 10,000 in Sweden. VP is particularly frequent in South Africa, where its high prevalence (>10,000 affected patients) is in part due to a genetic “founder effect.” The autosomal recessive acute hepatic porphyria, ALA dehydratase-deficient porphyria (ADP), is very rare, and less than 20 patients have been identified worldwide.
The porphyrias are panethnic metabolic diseases that affect individuals around the globe. The acute hepatic porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—are autosomal dominant disorders. The frequency of AIP, the most common acute hepatic porphyria, is ~1 in 20,000 among Caucasian individuals of Western European ancestry, and it is particularly frequent in Scandinavians, with a frequency of ~1 in 10,000 in Sweden. VP is particularly frequent in South Africa, where its high prevalence (>10,000 affected patients) is in part due to a genetic “founder effect.” The autosomal recessive acute hepatic porphyria, ALA dehydratase-deficient porphyria (ADP), is very rare, and less than 20 patients have been identified worldwide.
The erythropoietic protoporphyrias—CEP, erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP)—also are panethnic. EPP is the most common porphyria in children, whereas CEP is very rare, with about 200 reported cases worldwide. The frequency of EPP varies globally because most patients have the common low expression FECH mutation that varies in frequency in different populations. It rarely occurs in Africans, is present in about 10% of whites, and is frequent (~30%) in the Japanese.
The autosomal recessive porphyrias—ADP, CEP, EPP, and hepatoerythropoietic porphyria (HEP)—are more frequent in regions with high rates of consanguineous unions. PCT, which is typically sporadic, occurs more frequently in countries in which its predisposing risk factors such as hepatitis C and HIV are more prevalent.
HEME BIOSYNTHESIS
Heme biosynthesis involves eight enzymatic steps in the conversion of glycine and succinyl-CoA to heme (Fig. 430-2 and Table 430-2). These eight enzymes are encoded by nine genes, as the first enzyme in the pathway, 5′-aminolevulinate synthase (ALA synthase), has two genes that encode unique housekeeping (ALAS1) and erythroid-specific (ALAS2) isozymes. The first and last three enzymes in the pathway are located in the mitochondrion, whereas the other four are in the cytosol. Heme is required for a variety of hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, and the cytochrome P450 enzymes (CYPs). Hemoglobin synthesis in erythroid precursor cells accounts for approximately 85% of daily heme synthesis in humans. Hepatocytes account for most of the rest, primarily for the synthesis of CYPs, which are especially abundant in the liver endoplasmic reticulum, and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes. As shown in Fig. 430-2, pathway intermediates are the porphyrin precursors, ALA and PBG, and porphyrins (mostly in their reduced forms, known as porphyrinogens). At least in humans, these intermediates do not accumulate in significant amounts under normal conditions or have important physiologic functions.
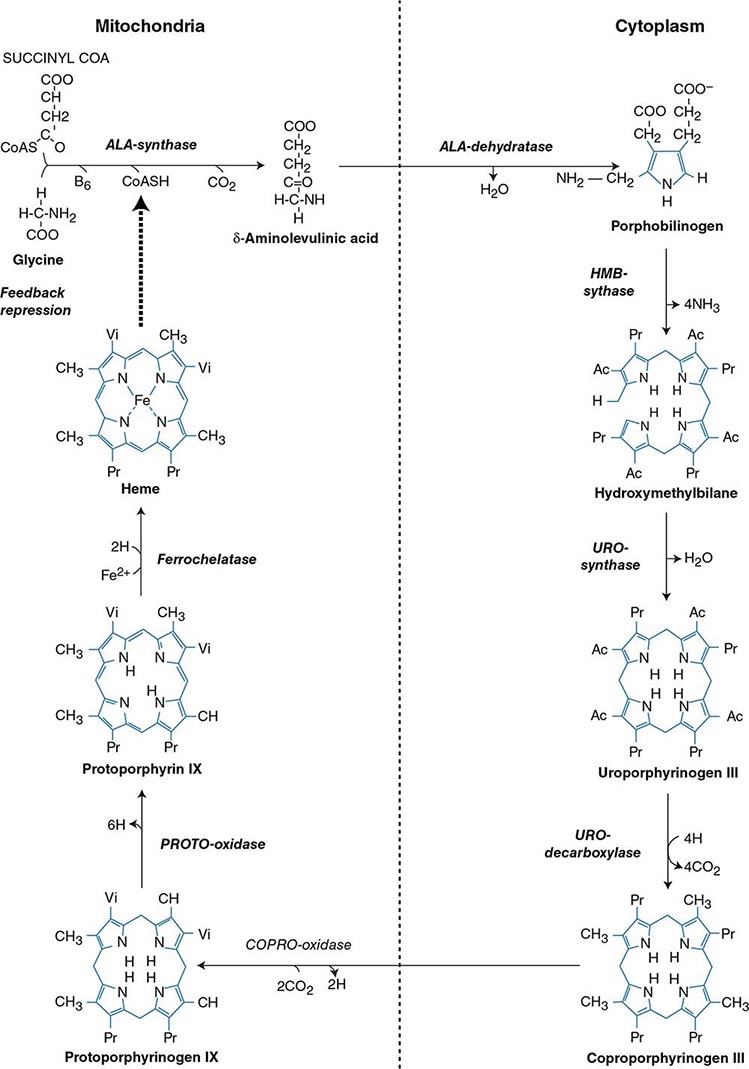
FIGURE 430-2 The heme biosynthetic pathway showing the eight enzymes and their substrates and products. Four of the enzymes are localized in the mitochondria and four in the cytosol.
The first enzyme, ALA synthase, catalyzes the condensation of glycine, activated by pyridoxal phosphate and succinyl coenzyme A, to form ALA. In the liver, this rate-limiting enzyme can be induced by a variety of drugs, steroids, and other chemicals. Distinct nonerythroid (e.g., housekeeping) and erythroid-specific forms of ALA synthase are encoded by separate genes located on chromosome 3p21.1 (ALAS1) and Xp11.2 (ALAS2), respectively. Defects in the erythroid gene ALAS2 that decrease its activity cause X-linked sideroblastic anemia (XLSA). Recently, gain-of-function mutations in the last exon (11) of ALAS2 that increase its activity have been shown to cause an X-linked form of EPP, known as X-linked protoporphyria (XLP).
The second enzyme, ALA dehydratase, catalyzes the condensation of two molecules of ALA to form PBG. Hydroxymethylbilane synthase (HMB synthase; also known as PBG deaminase) catalyzes the head-to-tail condensation of four PBG molecules by a series of deaminations to form the linear tetrapyrrole, HMB. Uroporphyrinogen III synthase (URO synthase) catalyzes the rearrangement and rapid cyclization of HMB to form the asymmetric, physiologic, octacarboxylate porphyrinogen, uroporphyrinogen (URO’gen) III.
The fifth enzyme in the pathway, uroporphyrinogen decarboxylase (URO decarboxylase), catalyzes the sequential removal of the four carboxyl groups from the acetic acid side chains of URO’gen III to form coproporphyrinogen (COPRO’gen) III, a tetracarboxylate porphyrinogen. This compound then enters the mitochondrion via a specific transporter, ABCB6, where COPRO oxidase, the sixth enzyme, catalyzes the decarboxylation of two of the four propionic acid groups to form the two vinyl groups of protoporphyrinogen (PROTO’gen) IX, a decarboxylate porphyrinogen. Next, PROTO oxidase oxidizes PROTO gen to protoporphyrin IX by the removal of six hydrogen atoms. The product of the reaction is a porphyrin (oxidized form), in contrast to the preceding tetrapyrrole intermediates, which are porphyrinogens (reduced forms). Finally, ferrous iron is inserted into protoporphyrin to form heme, a reaction catalyzed by the eighth enzyme in the pathway, ferrochelatase (also known as heme synthetase or protoheme ferrolyase).
REGULATION OF HEME BIOSYNTHESIS
Regulation of heme synthesis differs in the two major heme-forming tissues, the liver and erythron. In the liver, the concentration of “free” heme regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA synthase 1. Heme represses the synthesis of the ALA synthase 1 mRNA and interferes with the transport of the enzyme from the cytosol into mitochondria. Hepatic ALA synthase 1 is increased by many of the same chemicals that induce the cytochrome P450 enzymes in the endoplasmic reticulum of the liver. Because most of the heme in the liver is used for the synthesis of cytochrome P450 enzymes, hepatic ALA synthase 1 and the cytochrome P450s are regulated in a coordinated fashion, and many drugs that induce hepatic ALA synthase 1 also induce the CYP genes. The other hepatic heme biosynthetic enzymes are presumably expressed at constant levels, although their relative activities and kinetic properties differ. For example, normal individuals have high activities of ALA dehydratase, but low activities of HMB synthase, the latter being the second rate-limiting step in the pathway.
In the erythron, novel regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis. The response to stimuli for hemoglobin synthesis occurs during cell differentiation, leading to an increase in cell number. In contrast, the erythroid-specific ALA synthase 2 is expressed at higher levels than the housekeeping enzyme, and erythroid-specific control mechanisms regulate other pathway enzymes as well as iron transport into erythroid cells. Separate erythroid-specific and nonerythroid or “housekeeping” transcripts are known for the first four enzymes in the pathway. As noted above, housekeeping and erythroid-specific ALA synthases are encoded by genes on different chromosomes, but for each of the next three genes in the pathway, both erythroid and nonerythroid transcripts are transcribed by alternative promoters from their single respective genes (Table 430-2).
CLASSIFICATION OF THE PORPHYRIAS
As mentioned above, the porphyrias can be classified as either hepatic or erythropoietic, depending on whether the heme biosynthetic intermediates that accumulate arise initially from the liver or developing erythrocytes, or as acute or cutaneous, based on their clinical manifestations. Table 430-1 lists the porphyrias, their principal symptoms, and major biochemical abnormalities. Four of the five hepatic porphyrias—AIP, HCP, VP, and ADP—present during adult life with acute attacks of neurologic manifestations and elevated levels of one or both of the porphyrin precursors, ALA and PBG, and are thus classified as acute porphyrias. Patients with ADP have presented in infancy and adolescence. The fifth hepatic disorder, PCT, presents with blistering skin lesions. HCP and VP also may have cutaneous manifestations similar to PCT.
The erythropoietic porphyrias—CEP, EPP, and the recently described XLP—are characterized by elevations of porphyrins in bone marrow and erythrocytes and present with cutaneous photosensitivity. The skin lesions in CEP resemble PCT but are usually much more severe, whereas EPP and XLP cause a more immediate, painful, and nonblistering type of photosensitivity. EPP is the most common porphyria to cause symptoms before puberty. Around 20% of EPP patients develop minor abnormalities of liver function, with up to about 5% developing hepatic complications that can become life-threatening. XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
DIAGNOSIS OF PORPHYRIA
A few specific and sensitive first-line laboratory tests should be used whenever symptoms or signs suggest the diagnosis of porphyria (Table 430-3). If a first-line test is significantly abnormal, more comprehensive testing should follow to establish the type of porphyria, including the specific causative gene mutation.
Acute Porphyrias An acute porphyria should be suspected in patients with neurovisceral symptoms after puberty, such as abdominal pain, and when the initial clinical evaluation does not suggest another cause. The urinary porphyrin precursors (ALA and PBG) should be measured (Fig. 430-2). Urinary PBG is virtually always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition. Therefore, this measurement is both sensitive and specific. A method for rapid, in-house testing for urinary PBG, such as the Trace PBG kit (Thermo Scientific), can be used. Results from spot (single-void) urine specimens are highly informative because very substantial increases in PBG are expected during acute attacks of porphyria. A 24-h collection can unnecessarily delay diagnosis. The same spot urine specimen should be saved for quantitative determination of ALA, PBG, and creatinine, in order to confirm the qualitative PBG result and also to detect patients with ADP. Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP. Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific. Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance. Measurement of erythrocyte HMB synthase is not useful as a first-line test. Moreover, the enzyme activity is not decreased in all AIP patients, a borderline low normal value is not diagnostic, and the enzyme is not deficient in other acute porphyrias.
Cutaneous Porphyrias Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins. A fluorometric method is preferred, because the porphyrins in plasma in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-performance liquid chromatography (HPLC). The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease.
Although a total plasma porphyrin determination will usually detect EPP and XLP, an erythrocyte protoporphyrin determination is more sensitive. Increases in erythrocyte protoporphyrin occur in many other conditions. Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin. In XLP, both free and zinc protoporphyrin are markedly increased in approximately equal proportions. Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
More extensive testing is justified when an initial test is positive. A substantial increase in PBG may be due to AIP, HCP, or VP. These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins. Assays for COPRO oxidase or PROTO oxidase are not widely available. More specifically, mutation analysis by sequencing the genes encoding HMB synthase, COPRO oxidase, and PROTO oxidase will detect almost all disease-causing mutations, and will be diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal. The various porphyrias that cause blistering skin lesions are differentiated by measuring porphyrins in urine, feces, and plasma. These porphyrias also should be confirmed at the DNA level by the demonstration of the causative gene mutation(s). It is often difficult to diagnose or “rule out” porphyria in patients who have had suggestive symptoms months or years in the past, and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal. In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives. Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
THE HEPATIC PORPHYRIAS
Markedly elevated plasma and urinary concentrations of the porphyrin precursors, ALA and/or PBG, which originate from the liver, are especially evident during attacks of neurologic manifestations of the four acute porphyrias—ADP, AIP, HCP, and VP. In PCT, excess porphyrins also accumulate initially in the liver and cause chronic blistering of sun-exposed areas of the skin.
ALA DEHYDRATASE-DEFICIENT PORPHYRIA (ADP)
ADP is a rare autosomal recessive acute hepatic porphyria caused by a severe deficiency of ALA dehydratase activity. To date, there are only a few documented cases, some in children or young adults, in which specific gene mutations have been identified. These affected homozygotes had <10% of normal ALA dehydratase activity in erythrocytes, but their clinically asymptomatic parents and heterozygous relatives had about half-normal levels of activity and did not excrete increased levels of ALA. The frequency of ADP is unknown, but the frequency of heterozygous individuals with <50% normal ALA dehydratase activity was ~2% in a screening study in Sweden. Because there are multiple causes for deficient ALA dehydratase activity, it is important to confirm the diagnosis of ADP by mutation analysis.
Clinical Features The clinical presentation depends on the amount of residual ALA dehydratase activity. Four of the documented patients were male adolescents with symptoms resembling those of AIP, including abdominal pain and neuropathy. One patient was an infant with more severe disease, including failure to thrive beginning at birth. The earlier age of onset and more severe manifestations in this patient reflect a more significant deficiency of ALA dehydratase activity. Another patient developed an acute motor polyneuropathy at age 63 that was associated with a myeloproliferative disorder. He was heterozygous for an ALAD mutation that presumably was present in erythroblasts that underwent clonal expansion due to the bone marrow malignancy.
Diagnosis All patients had significantly elevated levels of plasma and urinary ALA and urinary coproporphyrin (COPRO) III; ALAD activities in erythrocytes were <10% of normal. Hereditary tyrosinemia type 1 (fumarylacetoacetase deficiency) and lead intoxication should be considered in the differential diagnosis because either succinylacetone (which accumulates in hereditary tyrosinemia and is structurally similar to ALA) or lead can inhibit ALA dehydratase, increase urinary excretion of ALA and COPRO III, and cause manifestations that resemble those of the acute porphyrias. Heterozygotes are clinically asymptomatic and do not excrete increased levels of ALA but can be detected by demonstration of intermediate levels of erythrocyte ALA dehydratase activity or a specific mutation in the ALAD gene. To date, molecular studies of ADP patients have identified nine point mutations, two splice-site mutations, and a two-base deletion in the ALAD gene (Human Gene Mutation Database; www.hgmd.org). The parents in each case were not consanguineous, and the index cases had inherited a different ALAD mutation from each parent. Prenatal diagnosis of this disorder is possible by determination of ALA dehydratase activity and/or gene mutations in cultured chorionic villi or amniocytes.
TREATMENT | ALA-DEHYDRATASE DEFICIENT PORPHYRIA |
The treatment of ADP acute attacks is similar to that of AIP (see below). The severely affected infant referred to above was supported by hyperalimentation and periodic blood transfusions but did not respond to intravenous hemin and died after liver transplantation.
ACUTE INTERMITTENT PORPHYRIA (AIP)
This hepatic porphyria is an autosomal dominant condition resulting from the half-normal level of HMB synthase activity. The disease is widespread but is especially common in Scandinavia and Great Britain. Clinical expression is highly variable, and activation of the disease is often related to environmental or hormonal factors, such as drugs, diet, and steroid hormones. Attacks can be prevented by avoiding known precipitating factors. Rare homozygous dominant AIP also has been described in children (see below).
Clinical Features Induction of the rate-limiting hepatic enzyme ALA synthase in heterozygotes who have half-normal HMB synthase activity is thought to underlie the acute attacks in AIP. The disorder remains latent (or asymptomatic) in the great majority of those who are heterozygous for HMBS mutations, and this is almost always the case prior to puberty. In patients with no history of acute symptoms, porphyrin precursor excretion is usually normal, suggesting that half-normal hepatic HMB synthase activity is sufficient and that hepatic ALA synthase activity is not increased. However, under conditions where heme synthesis is increased in the liver, half-normal HMB synthase activity may become limiting, and ALA, PBG, and other heme pathway intermediates may accumulate and be excreted in the urine. Common precipitating factors include endogenous and exogenous steroids, porphyrinogenic drugs, alcohol ingestion, and low-calorie diets, usually instituted for weight loss.
The fact that AIP is almost always latent before puberty suggests that adult levels of steroid hormones are important for clinical expression. Symptoms are more common in women, suggesting a role for estrogens or progestins. Premenstrual attacks are probably due to endogenous progesterone. Acute porphyrias are sometimes exacerbated by exogenous steroids, including oral contraceptive preparations containing progestins. Surprisingly, pregnancy is usually well tolerated, suggesting that beneficial metabolic changes may ameliorate the effects of high levels of progesterone. Table 430-4 provides a partial list of the major drugs that are harmful in AIP (and also in HCP and VP). Extensive lists of unsafe and safe drugs are available on websites sponsored by the American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Initiative (www.porphyria-europe.org), and at the Drug Database for Acute Porphyrias website (www.drugs-porphyria.com). Reduced intake of calories and carbohydrate, as may occur with illness or attempts to lose weight, can also increase porphyrin precursor excretion and induce attacks of porphyria. Increased carbohydrate intake may ameliorate attacks. Studies in a knockout AIP mouse model indicate that the hepatic ALAS1 gene is regulated by the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Hepatic PGC-1α is induced by fasting, which in turn activates ALAS1 transcription, resulting in increased heme biosynthesis. This finding suggests an important link between nutritional status and the attacks in acute porphyrias. Attacks also can be provoked by infections, surgery, and ethanol.
UNSAFE DRUGS IN PORPHYRIA |
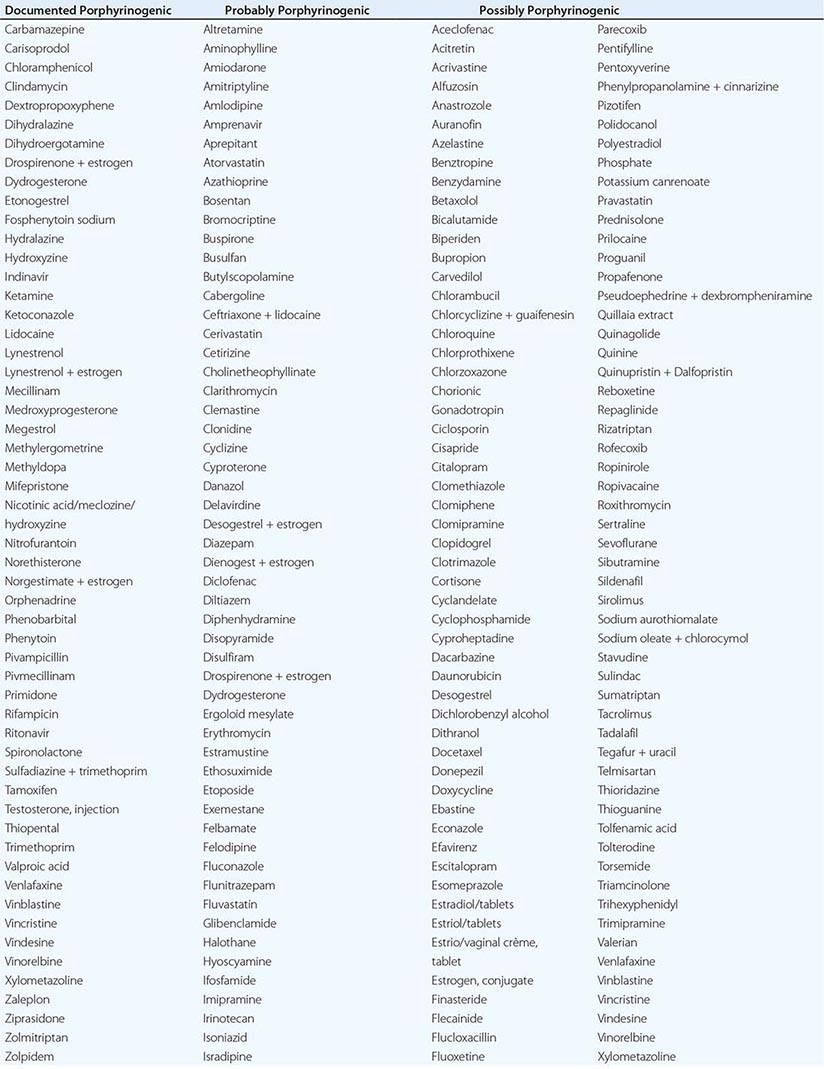

Because the neurovisceral symptoms rarely occur before puberty and are often nonspecific, a high index of suspicion is required to make the diagnosis. The disease can be disabling but is rarely fatal. Abdominal pain, the most common symptom, is usually steady and poorly localized but may be cramping. Ileus, abdominal distention, and decreased bowel sounds are common. However, increased bowel sounds and diarrhea may occur. Abdominal tenderness, fever, and leukocytosis are usually absent or mild because the symptoms are neurologic rather than inflammatory. Nausea; vomiting; constipation; tachycardia; hypertension; mental symptoms; pain in the limbs, head, neck, or chest; muscle weakness; sensory loss; dysuria; and urinary retention are characteristic. Tachycardia, hypertension, restlessness, tremors, and excess sweating are due to sympathetic overactivity.
The peripheral neuropathy is due to axonal degeneration (rather than demyelinization) and primarily affects motor neurons. Significant neuropathy does not occur with all acute attacks; abdominal symptoms are usually more prominent. Motor neuropathy affects the proximal muscles initially, more often in the shoulders and arms. The course and degree of involvement are variable and sometimes may be focal and involve cranial nerves. Deep tendon reflexes initially may be normal or hyperactive but become decreased or absent as the neuropathy advances. Sensory changes such as paresthesia and loss of sensation are less prominent. Progression to respiratory and bulbar paralysis and death occurs especially when the diagnosis and treatment are delayed. Sudden death may result from sympathetic overactivity and cardiac arrhythmia.
Mental symptoms such as anxiety, insomnia, depression, disorientation, hallucinations, and paranoia can occur in acute attacks. Seizures can be due to neurologic effects or to hyponatremia. Treatment of seizures is difficult because most antiseizure drugs can exacerbate AIP (clonazepam may be safer than phenytoin or barbiturates). Hyponatremia results from hypothalamic involvement and inappropriate vasopressin secretion or from electrolyte depletion due to vomiting, diarrhea, poor intake, or excess renal sodium loss. Persistent hypertension and impaired renal function may occur. When an attack resolves, abdominal pain may disappear within hours, and paresis begins to improve within days and may continue to improve over several years.
Homozygous dominant AIP is a rare form of AIP in which patients inherit HMBS mutations from each of their heterozygous parents and, therefore, have very low (<2%) enzyme activity. The disease has been described in a Dutch girl, two young British siblings, and a Spanish boy. In these homozygous affected patients, the disease presented in infancy with failure to thrive, developmental delay, bilateral cataracts, and/or hepatosplenomegaly. Urinary ALA and PBG concentrations were markedly elevated. All of these patients’ HMBS mutations (R167W, R167Q, and R172Q) were in exon 10 within five bases of each other. Studies of the brain magnetic resonance images (MRIs) of children with homozygous AIP have suggested damage primarily in white matter that was myelinated postnatally, while tracks that myelinated prenatally were normal. Most children with homozygous AIP die at an early age.
Diagnosis ALA and PBG levels are substantially increased in plasma and urine, especially during acute attacks, and become normal only after prolonged latency. For example, urinary PBG excretion during an attack is usually 50–200 mg/24 h (220–880 μmol/24 h) (normal, 0–4 mg/24 h, [0–18 μmol/24 h]), and urinary ALA excretion is 20–100 mg/24 h (150–760 μmol/24 h) (normal, 1–7 mg/24 h [8–53 μmol/24 h]). Because levels often remain high after symptoms resolve, the diagnosis of an acute attack in a patient with biochemically proven AIP is based primarily on clinical features. Excretion of ALA and PBG decreases over a few days after intravenous hemin administration. A normal urinary PBG level before hemin effectively excludes AIP as a cause for current symptoms. Fecal porphyrins are usually normal or minimally increased in AIP, in contrast to HCP and VP. Most AIP heterozygotes with no history of symptoms have normal urinary excretion of ALA and PBG. Therefore, the detection of the family’s HMBS mutation will diagnose asymptomatic family members.
Patients with HMBS mutations in the initiation of translation codon in exon 1 and in the intron 15′-splice donor site have normal enzyme levels in erythrocytes and deficient activity only in nonerythroid tissues. This occurs because the erythroid and housekeeping forms of HMB synthase are encoded by a single gene, which has two promoters. Thus, the enzyme assay may not be diagnostic, and genetic testing should be used to confirm the diagnosis.
More than 390 HMBS mutations have been identified in AIP, including missense, nonsense, and splicing mutations and insertions and deletions, with most mutations found in only one or a few families (Human Gene Mutation Database, www.hgmd.org). The prenatal diagnosis of a fetus at risk can be made with cultured amniotic cells or chorionic villi. However, this is seldom done, because the prognosis of individuals with HMBS mutations is generally favorable.
TREATMENT | ACUTE INTERMITTENT PORPHYRIA |
During acute attacks, narcotic analgesics may be required for abdominal pain, and phenothiazines are useful for nausea, vomiting, anxiety, and restlessness. Chloral hydrate can be given for insomnia, and benzodiazepines are probably safe in low doses if a minor tranquilizer is required. Carbohydrate loading, usually with intravenous glucose (at least 300 g daily), may be effective in milder acute attacks of porphyria (without paresis, hyponatremia, etc.) if hemin is not available. Intravenous hemin is more effective and should be used as first-line therapy for all acute attacks. The standard regimen is 3–4 mg/kg of heme, in the form of lyophilized hematin (Recordati Pharmaceuticals), heme albumin (hematin reconstituted with human albumin), or heme arginate (Orphan Europe), infused daily for 4 days. Heme arginate and heme albumin are chemically stable and are less likely than hematin to produce phlebitis or an anticoagulant effect. Recovery depends on the degree of neuronal damage and usually is rapid if therapy is started early. Recovery from severe motor neuropathy may require months or years. Identification and avoidance of inciting factors can hasten recovery from an attack and prevent future attacks. Inciting factors are usually multiple, and removal of one or more hastens recovery and helps prevent future attacks. Frequent attacks that occur during the luteal phase of the menstrual cycle may be prevented with a gonadotropin-releasing hormone analogue, which prevents ovulation and progesterone production, or by prophylactic hematin administration.
The long-term risk of hypertension and chronic renal disease is increased in AIP; a number of patients have undergone successful renal transplantation. Chronic, low-grade abnormalities in liver function tests are common, and the risk of hepatocellular carcinoma is increased. Hepatic imaging is recommended at least yearly for early detection of these tumors.
An allogeneic liver transplant was performed on a 19-year-old female AIP heterozygote who had 37 acute attacks in the 29 months prior to transplantation. After transplantation, her elevated urinary ALA and BPG levels returned to normal in 24 h, and she did not experience acute neurologic attacks for more than 3 years after transplant. Two AIP patients had combined liver and kidney transplants secondary to uncontrolled acute porphyria attacks, chronic peripheral neuropathy, and renal failure requiring dialysis. Both patients had a marked improvement with no attacks and normal urinary PBG levels after transplantation, as well as improvement of their neuropathic manifestations. More recently, a group from the United Kingdom reported their experience with liver transplantation in 10 AIP patients with recurrent attacks that were refractory to medical management and impaired quality of life. Patients had a complete biochemical and symptomatic resolution after transplant. The investigators reported a high rate of hepatic artery thrombosis in their series. Clearly, liver transplantation is a high-risk procedure and should be considered as a last resort in patients with severe recurrent attacks. Recently, liver-directed gene therapy has proven successful in the prevention of drug-induced biochemical attacks in a murine model of human AIP, and clinical trials of AAV-HMBS gene transfer have been initiated. In addition, preclinical studies of a hepatic-targeted RNA interference (RNAi) therapy directed to inhibit the markedly elevated hepatic ALAS1 mRNA in the AIP mouse model prevented induced biochemical attacks and rapidly reduced the ALAS1 mRNA during an ongoing attack.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


