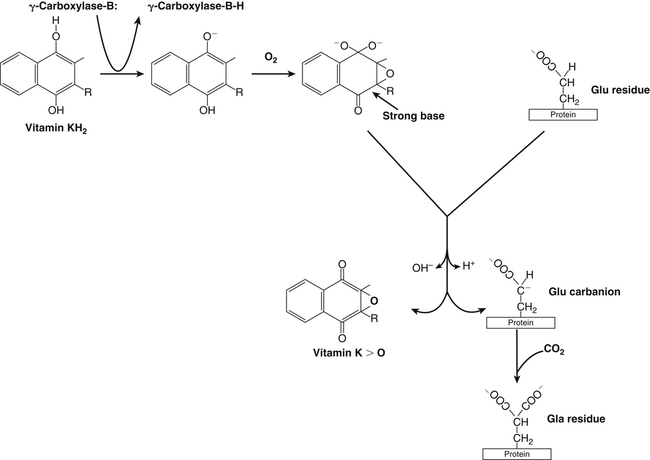
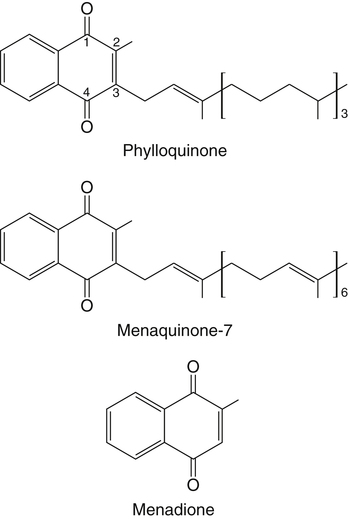
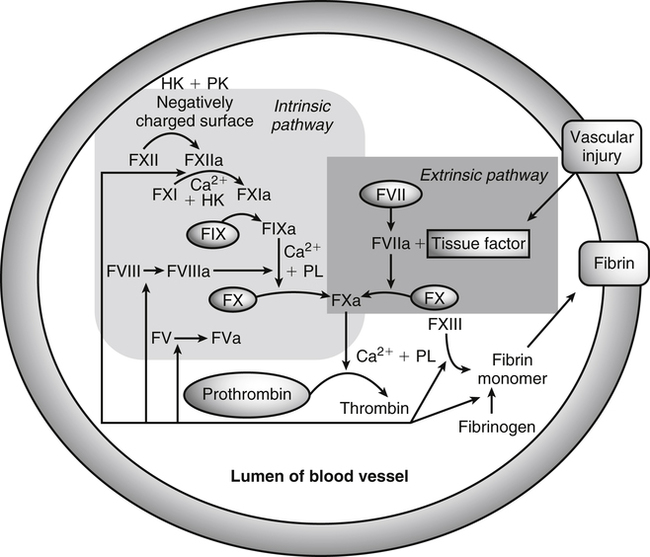
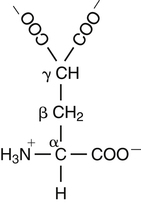
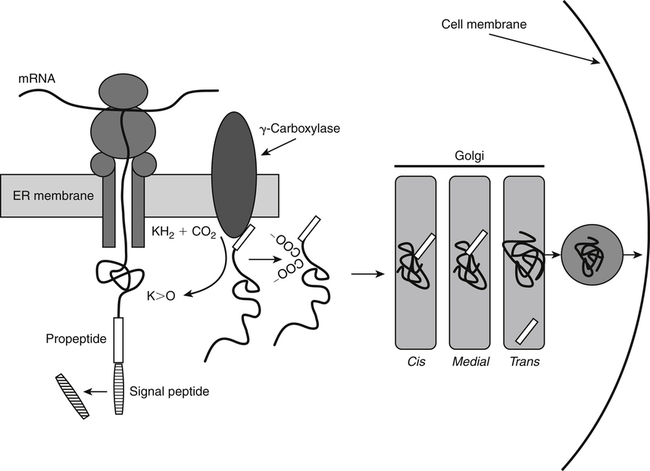
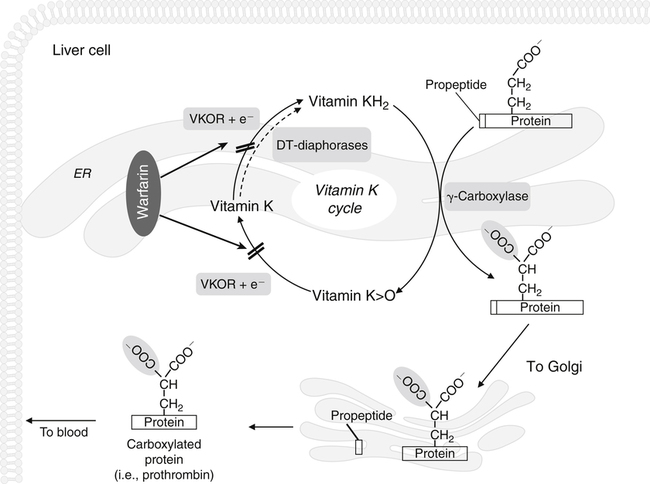
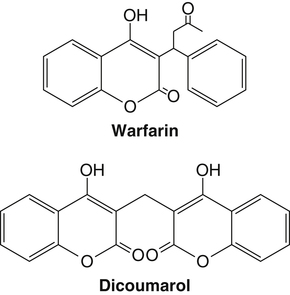
Vitamin K, a fat-soluble vitamin, was first identified as an antihemorrhagic factor in the late 1930s (Dam, 1934) and is so named because a lack of this vitamin causes a defect in blood koagulation (German word for coagulation). Vitamin K serves as a cofactor in a posttranslational modification reaction converting a specific set of glutamic acid (Glu) residues in precursors of vitamin K–dependent proteins to γ-carboxyglutamic acid (Gla) residues which can bind Ca2+. The vitamin K–dependent modification system is located on the luminal side of the endoplasmic reticulum (ER) where newly synthesized proteins undergo folding and modifications (Stanton et al., 1991). The first proteins to be characterized as vitamin K–dependent proteins were those belonging to the blood coagulation system (Monroe et al., 2002). The independent discovery by Stenflo and colleagues (1974) and Nelsestuen and co-workers (1974) that the vitamin K–dependent modification was a postribosomal modification converting Glu residues to Ca2+-binding Gla residues in newly synthesized vitamin K–dependent proteins yielded the clinically important understanding as to why Ca2+ is essential for hemostasis. Ca2+ binding by the clotting factors, prothrombin and factors VII, IX and X, is essential for assembly of coagulation complexes on phospholipids at the site of an injury in the vessel wall, where blood platelets are first to aggregate and provide the phospholipids. Following the discovery of vitamin K–dependent proteins belonging to the coagulation system, several vitamin K–dependent proteins that do not belong to the coagulation system have been identified, including matrix Gla protein (MGP) involved in inhibition of pathological bone formation; osteocalcin, involved in bone metabolism; and Gas6, involved in cell survival (Booth, 2009). A role for vitamin K in regulation of calcification, energy metabolism, and inflammation has been suggested (Booth, 2009). Compounds with vitamin K activity are 2-methyl-1,4-naphthoquinones with an isoprenoid hydrophobic side chain at the 3 position (Figure 28-1). Phylloquinone, also known as vitamin K1, is the form of vitamin K that is isolated from green plants. Phylloquinone has a phytyl group at the 3 position of the naphthoquinone ring. The vitamin K2 forms of the vitamin that are synthesized by bacteria are now more properly designated as menaquinones. These forms of the vitamin have a side chain composed of unsaturated isoprene units at the 3 position (see Figure 28-1). Although a wide range of menaquinones (abbreviated as MK-n) are synthesized by bacteria, long-chain menaquinones with 6 to 10 isoprene groups in the side chain (MK-6 to MK-10) are the most common. The synthetic compound menadione (2-methyl-1,4-naphthoquinone) (see Figure 28-1) was shown very early to have vitamin K activity and commonly is used as a source of the vitamin in animal feeds. Menadione itself is not a substrate for the vitamin K–dependent carboxylase, but it is alkylated in mammalian tissues to an active form, MK-4, having a geranylgeranyl side chain in the 3 position. The discovery of vitamin K as an antihemorrhagic factor directed focus on the blood coagulation system as the physiological system targeted by the vitamin. The initial models of the system proposed by Macfarlane (1965) and Davie and Ratnof (1964) involve a cascade of activations of blood coagulation factors that result in conversion of the soluble plasma protein fibrinogen into fibrin, a protein that aids formation of blood clots. These early models have been modified extensively, and a simplified version of the current model of the blood coagulation system leading to fibrin clot formation is shown in Figure 28-2. The four vitamin K–dependent clotting factors (prothrombin and factors VII, IX, and X) involved in the intrinsic and extrinsic pathways of the cascade are circled in Figure 28-2. These proteins were shown to bind calcium ions (Ca2+) and to bind to negatively charged phospholipid surfaces. The intrinsic and the extrinsic pathways of blood clotting converge at the formation of active factor Xa (FXa) from its inactive proenzyme factor X (FX) form. Studies of prothrombin production became the key to determining the mechanism of action of vitamin K. Experiments with whole animals carried out in the mid-1960s strongly suggested that vitamin K was involved in converting an inactive hepatic precursor of plasma prothrombin to a biologically active prothrombin form found in plasma. This hypothesis was strengthened by clinical observations that an immunochemically similar, but biologically inactive, form of prothrombin was present in increased concentrations in the plasma of patients treated with anticoagulants that antagonized vitamin K action. The inactive proteins were called PIVKA factor (proteins induced in vitamin K absence) (Hemker and Reekers, 1974). Characterization of this “abnormal prothrombin” isolated from the plasma of cows fed the anticoagulant dicoumarol revealed that it lacked the specific Ca2+-binding sites present in normal prothrombin, and it did not demonstrate a Ca2+-dependent association with negatively charged phospholipid surfaces. These findings suggested that the function of vitamin K was to modify a liver precursor of this plasma protein to facilitate calcium binding. The action of the vitamin was therefore not at the level of gene transcription, but occurred after messenger RNA (mRNA) translation. By comparing peptides obtained from proteolytic digests of normal bovine prothrombin and abnormal prothrombin obtained from cows treated with dicumarol, Stenflo and colleagues (1974) identified the vitamin K modification as γ-carboxylation of Glu residues in newly formed vitamin K–dependent protein precursors. The structure of Gla, a previously unrecognized acidic amino acid (Nelsestuen et al., 1974; Stenflo et al., 1974), is shown in Figure 28-3. It was found that that all 10 of the Glu residues in the first 42 residues of bovine prothrombin were converted to Gla. The discovery of Gla in vitamin K–dependent coagulation factors revealed, for the first time, the role of Ca2+ in blood clotting. Calcium-binding by the Gla residues is essential for anchoring these coagulation factors to negatively charged lipids at the site of injury (Furie and Furie, 1992). The function of Gla residues in the vitamin K–dependent proteins that do not belong to the blood clotting system is less understood. The discovery of Gla residues in prothrombin led to the demonstration (Esmon et al., 1975) that crude rat liver microsomal preparations (vesicles derived from the ER when cells are disrupted) contained an enzymatic activity that promoted a vitamin K–dependent incorporation of 14CO2 into endogenous precursors of vitamin K–dependent proteins present in these preparations. Subsequent studies established that the 14CO2 was present in Gla residues. Small peptides containing adjacent Glu-Glu residues such as Phe-Leu-Glu-Glu-Val were shown to be substrates for the enzyme present in detergent-solubilized microsomal preparations, and they were used to study the properties of this unique vitamin K–dependent γ-carboxylase. It is now well established that vitamin K–dependent γ-carboxylation of Glu residues in proteins is a posttranslational modification reaction that occurs on the luminal side of the rough ER. The majority of vitamin K–dependent proteins are secretory proteins that migrate through the ER and the Golgi apparatus before the proteins are secreted from the cells as mature proteins (Stanton et al., 1991). As shown in Figure 28-4, the vitamin K–dependent proteins are equipped with a signal peptide for penetration into the ER lumen. Upon entering the lumen, the N-terminal signal peptide is removed by signal peptidase. In addition, all precursors of vitamin K–dependent proteins are equipped with a unique propeptide that, in most of the proteins, constitutes the N-terminal part of the precursors after the signal peptide is removed. These propeptides are recognized by the γ-carboxylase in the ER membrane. They anchor the vitamin K–dependent protein precursors to the enzyme for γ-carboxylation of Glu residues in a reaction that requires reduced vitamin K (KH2) as the active cofactor. Upon completion of γ-carboxylation, the vitamin K–dependent proteins are released from the γ-carboxylase and exit the ER in secretory vesicles for transport to the Golgi apparatus for additional modifications. The major modifications these proteins undergo in the Golgi apparatus are glycosylations. The proteins move through the cis-, medial-, and trans-Golgi compartments (see Figure 28-4). Because of the addition of sialic acid residues, the vitamin K–dependent proteins become more acidic in the Golgi apparatus. The propeptide stays attached to the protein during its migration through the Golgi apparatus but is proteolytically released from the protein in the trans-Golgi apparatus by the protease furin (Stanton et al., 1991). Release of the propeptide is essential for the coagulation factors to function as zymogens in the blood coagulation system. Bleeding disorders have been shown to result in the appearance of vitamin K–dependent coagulation factors in blood with the propeptide still attached (Diuguid et al., 1986). As indicated in Figure 28-5, reduced vitamin K (KH2 or vitamin K hydroquinone) is the γ-carboxylase cofactor. When one Glu residue is converted to one Gla residue, the cofactor KH2 is converted to vitamin K 2,3-epoxide (K>O) (see Figure 28-5). The epoxide (K>O) is reduced back to the hydroquinone (KH2) cofactor by the enzyme vitamin K 2,3-epoxide reductase (VKOR). VKOR is inhibited by warfarin, a derivative of the anticoagulant drug dicoumarol. The oxidation of vitamin KH2 in the γ-carboxylation reaction and the reduction of K>O back to KH2 by VKOR constitute the vitamin K cycle. DT-diaphorases, enzymes that are able to use either NADH or NADPH, can reduce vitamin K to KH2 but cannot reduce K>O (see Figure 28-5). The warfarin-insensitive DT-diaphorase pathway for vitamin K reduction has a high Km for vitamin K1 and can only produce enough KH2 cofactor for the γ-carboxylase to optimally produce fully γ-carboxylated proteins when high tissue concentrations of vitamin K are present (Wallin and Martin, 1987). It has been a challenge over the years to determine how vitamin KH2 can be the trigger for γ-carboxylation of specific Glu residues in vitamin K–dependent proteins. The first fundamental and critical role of vitamin KH2 to initiate γ-carboxylation is its conversion into a strong base by removal of a H+ from one of the OH groups on the napthoquinone ring system by a strong basic amino acid residue (B:) in the γ-carboxylase enzyme (Figure 28-6). The strong vitamin K base (K−) thus formed then extracts a H+ at the γ-carbon of the targeted Glu residue in the protein, and the resulting carbanion is then attacked by CO2, resulting in addition of an extra COOH group to the γ-carbon on the Glu residue. Thus, a new Ca2+-binding Gla residue forms in the protein sequence. For a more detailed understanding of the chemical reaction modeling, the reader is referred to papers by Dowd and co-workers (1995) and by Rishavy and associates (2006). The physiological role of the vitamin K–dependent γ-carboxylase poses an interesting question in terms of enzyme–substrate recognition. This microsomal (ER) enzyme recognizes a small fraction of the total hepatic secretory protein pool and then carboxylates 9 to 12 Glu sites in the first 45 N-terminal residues of the vitamin K–dependent plasma proteins. The propeptide regions of these proteins show sequence homology. The propeptide region appears to be both a docking and a recognition site for the γ-carboxylase; it modulates the activity by decreasing the apparent Km of the γ-carboxylase for the Glu-containing protein substrates. The propeptide domain is undoubtedly of major importance in directing the efficient γ-carboxylation of the multiple Glu sites in these substrates, but it is not known if the enzyme starts at one end of the Gla region and sequentially carboxylates all Glu sites or if the enzyme carboxylates randomly within this region. Reduction of KH2 levels by anticoagulant therapy results in the production of a complex mixture of partially carboxylated forms of prothrombin, and the available data suggest that in the least carboxylated forms the potential Gla residue that is closest to the amino-terminal is preferentially carboxylated (Malhotra, 1972). The molecular mechanism resulting in the appearance in blood of vitamin K–dependent, under–γ-carboxylated coagulation factors (PIVKA factors), when the γ-carboxylation system is not functioning optimally, is unknown. A model for Glu γ-carboxylation in the presence of sufficient KH2 cofactor for the γ-carboxylase has been proposed by Stenina and colleagues (2001). Their data are consistent with a model of tethered processivity of γ-carboxylation. Finished Gla residues are bound to the active γ-carboxylation site on the γ-carboxylase that exposes a new targeted Glu residue for γ-carboxylation. This process will continue until the Gla region is completed. The finished vitamin K–modified protein then leaves the γ-carboxylase. Early studies of γ-carboxylation were hampered by the lack of purified γ-carboxylase. By using an affinity approach, where the ligand was a large peptide containing the propeptide region and the Gla region of factor IX, Wu and colleagues (1991) purified a microsomal protein of molecular weight 94 kDa that exhibited γ-carboxylase activity and that required lipid for activity. Data from a topological study carried out by Tie and colleagues (2000) predict that γ-carboxylase spans the ER membrane at least five times. Therefore γ-carboxylase is considered a true integral protein of the ER membrane. For a more detailed description of γ-carboxylase, the reader should refer to the paper by Rishavy and co-workers (2006). Dicoumarol was isolated from moldy sweet clover and identified in 1939 as the factor responsible for a hemorrhagic disease of cattle that was common in the midwestern United States in the early 1920s. This led to the use of dicoumarol (marketed in the United States as dicumarol) as the first anticoagulant drug to be used clinically. However, the toxicity of dicoumarol led to synthesis of many new versions of 4-hydroxycoumarins to find a clinically safe drug, and warfarin (Figure 28-7), synthesized under the leadership of Dr. Karl P. Link at the University of Wisconsin, became the most successful anticoagulant drug. Warfarin is an indirect vitamin K antagonist in that it does not inhibit the γ-carboxylase (Hildebrandt and Suttie, 1982) but rather inhibits VKOR and therefore the regeneration of vitamin KH2 from K>O. As shown in Figure 28-5, the anticoagulant warfarin inhibits VKOR of the vitamin K cycle. Warfarin resistance is a problem in prophylactic medicine and also in rodent pest control. The problem can be pharmacological, nutritional, or genetic. Pharmacological resistance is associated with high turnover of the drug in the liver by the cytochrome P450 system where cytochrome P450-2C9 is the most active cytochrome involved in metabolism of warfarin (Hanatani et al., 2003). Nutritional resistance to warfarin results from high vitamin K intake (Suttie, 1978), and administration of vitamin K is the standard procedure used to treat warfarin intoxication in patients and animals. The mechanism by which vitamin K works as an antidote to warfarin has been established (Wallin and Martin, 1987). When the liver concentration of vitamin K is elevated, vitamin K can be reduced by DT-diaphorases, enzymes that are able to use either NADH or NADPH for vitamin K reduction (Wallin and Martin, 1987; see Figure 28-5). Warfarin does not inhibit these enzymes. Therefore if the VKOR pathway for vitamin K reduction is blocked by warfarin, the alternative pathway can “drive” vitamin KH2 cofactor production and maintain a normally functioning blood Warfarin and Pharmacogenetics The basis of warfarin resistance is less worked out than that for warfarin sensitivity. Several single-point mutations that result in amino acid substitutions have been associated with warfarin resistance. Among warfarin resistant patients in a Israeli Jewish population, Loebstein et al. (2007) identified a 5417G>T (Asp36Tyr) polymorphism in 7 of 15 resistant patients. This variant was significantly and consistently associated with the highest warfarin dose category. Analysis of ethnic groups within this population showed over-representation of the Asp36Tyr polymorphism in individuals of Ethiopian descent (15%) and Ashkenazi descent (4%). 1. Describe the mechanism by which warfarin decreases blood clot formation. 2. What does prothrombin time reveal? 3. Why was John asked about his use of vitamin K supplements? 4. From the following choices, select the type of genetic variant in the VKORC1 gene that may have contributed to John’s decreased sensitivity to warfarin (may be more than one possible answer). coagulation system as long as vitamin K levels are elevated. Because the DT-diaphorases cannot reduce K>O (vitamin K 2,3 epoxide), vitamin K cannot be effectively recycled for participation in a second carboxylation reaction. Genetic warfarin resistance has been described in humans (Vermeer and Schurgers, 2000; Voora et al., 2009), and resistance to some of the second-generation anticoagulants has been seen in rodents. Members of two families with familial multiple coagulation factor deficiency (FMFD), a disease in which patients are deficient in all vitamin K–dependent coagulation factors, were investigated by Fregin and colleagues (2002), who mapped the disease to the 16p12-q21 region on chromosome 16. They noted that a linkage group of genes on 16p11 is orthologous to genes around warfarin resistance loci Rw in rats (Greaves and Ayres, 1967; Kohn et al., 2003) and War in mice (Wallace and MacSwiney, 1976). This homology led the investigators to propose that FMFD and warfarin resistance may be caused by allelic mutations in the same gene. DNA sequencing of genes in the region resulted in identification of missense mutations in a gene of unknown function from all of the FMFD patients studied. The wild-type gene in unaffected individuals codes for an 18-kDa ER membrane protein, now known as VKOR complex subunit 1 (VKORC1); overexpression of this gene yielded a protein that exhibited warfarin-sensitive VKOR activity. Furthermore, it was shown that certain missense mutations in the gene resulted in significant warfarin resistance in the host (Rost et al., 2004). Using synthetic short interfering RNAs (siRNAs) to silence genes in the warfarin-resistant region on chromosome 16, Li and colleagues (2004) identified the same gene as that responsible for expression of warfarin-sensitive VKOR activity. It has been proposed that additional forms of genetic warfarin resistance exist. One form has been shown to be associated with overexpression of the chaperone protein calumenin in rat liver (Wallin et al., 2001a). Calumenin belongs to the CREC (Cab45, reticulocalbin, ERC-45, calumenin) subfamily of Ca2+-binding proteins (Honore, 2009; Honore and Vorum, 2000). The CREC proteins are found in the secretory pathways of cells, and most of the proteins have been shown to have chaperone functions. Despite being a hydrophilic, water-soluble, acidic protein, calumenin appears to be associated with ER membrane proteins. Data supporting this concept include immunocytochemical studies of the ER membrane (Honore and Vorum, 2000) and the demonstration that calumenin is strongly associated with lipid–detergent micelles, derived from the ER membrane, which also carry the VKOR enzyme complex (Wallin et al., 2001a). Expression of calumenin in cell lines has been shown to inhibit VKOR activity and confer warfarin resistance to the VKOR enzyme complex. In addition, calumenin has been shown to be associated with the γ-carboxylase and to inhibit its activity (Wajih et al., 2004). Therefore calumenin appears to be a chaperone that regulates the capacity of the vitamin K–dependent γ-carboxylation system for production of functional vitamin K–dependent proteins. This regulatory role puts calumenin in a central position regarding maintenance of a normal blood coagulation system. In a recent human genetic study by Voora and co-workers (2009) calumenin was found to be a marker for warfarin resistance in African Americans and to have a role in the γ-carboxylation system, as suggested earlier by Wallin and colleagues (2001a).
Vitamin K
Nomenclature of Vitamin K Active Compounds
Mechanism of Action of Vitamin K
Discovery of the Physiological Mechanism of Vitamin K Action
Vitamin K: A Substrate for a γ-Carboxylase
The Vitamin K Cycle and the Vitamin K–Dependent γ-Carboxylation System
Chemistry of Vitamin K Action
γ-Carboxylation of Protein Glu Residues
Purification and Characterization of γ-Carboxylase
Antagonism of Vitamin K Action by Clinically Used Inhibitors
Warfarin Resistance and the Vitamin K–Dependent γ-Carboxylation System
 CLINICAL CORRELATION
CLINICAL CORRELATION
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


