1Abbreviations: 1α,25-(OH)2D, 1α,25-dihydroxy vitamin D; 1α,25-(OH)2D3, 1α,25-dihydroxy vitamin D3; 25-OH-D, 25-hydroxyvitamin D; BMI, body mass index; Ca2+, calcium ion; CYP, cytochrome P-450; DBP, vitamin D-binding protein; DRI, dietary reference intake; FGF-23, fibroblast growth factor 23; IOM, Institute of Medicine; NHANES, National Health and Nutrition Examination Survey; PCR, polymerase chain reaction; PTH, parathyroid hormone; RDA, recommended dietary allowance; UV, ultraviolet; VDR, vitamin D receptor; VDRE, vitamin D-responsive element.
HISTORY OF RICKETS AND VITAMIN D AS AN ANTIRACHITIC FACTOR
Although vitamin D was discovered less than 100 years ago, its deficiency diseases (rickets, and its adult-onset counterpart, osteomalacia) were clearly recognized by physicians such as Daniel Whistler (1645) in the Netherlands and Francis Glisson (1650) in England much earlier (1, 2). Rickets is characterized by a “bone matrix which is insufficiently mineralized or calcified,” usually as a result of vitamin D deficiency. Rickets results in a deformed and misshaped skeleton, particularly bending or bowing of the long bones and enlargement of the epiphyses of the joints of the rib cage, arms, legs, and neck. These defects result in painful breathing and difficulties holding up the head, and in women they even give rise to problems with childbirth in later life, if rachitic deformities of the pelvis persist. By the turn of the twentieth century, rickets had reached almost epidemic proportions, particularly in the industrialized cities of Northern Europe, where the excessive degree of air pollution that blocked ultraviolet (UV) exposure and the sweatshop labor practices that kept workers indoors during the daylight hours combined to prevent skin synthesis of vitamin D.
During the nineteenth and early twentieth centuries, the following physicians and scientists investigated the cause of rickets and made key observations that led to the discovery of vitamin D: Sniadecki (1822), who noted that rickets was common in city-dwelling children but not rural dwellers (3); Palm (1890), who noted the importance of latitude in the incidence of rickets in China and concluded that sunshine is the main etiologic factor in rickets (4); Percival (1789), who discussed the medicinal uses of cod liver oil in treating rickets (5); Raczynski (1913), Huldschinsky (1919), and later Hess and Unger (1922), who showed that laboratory animals and children with rickets were cured when exposed to sunlight or mercury (UV) lamps (6, 7, 8); Mellanby (1919), who fed a semisynthetic oatmeal diet to dogs to induce rickets, which was reversible by administering cod liver oil (9); McCollum et al (1922), who demonstrated that the antirachitic factor in cod liver oil was distinct from vitamin A and named it vitamin D (10); Hess and Weinstock (1924) and Steenbock and Black (1924), who were able to reconcile the apparently disparate concepts of sunlight and dietary factors by showing that irradiation of certain food (e.g., plant oils or yeast) increased vitamin D activity (11, 12) (this plant-derived antirachitic factor is now known as vitamin D2); and Windaus et al (1936), who received the Nobel Prize in 1928 principally for elucidating the structure of sterols, including vitamin D (13).
Terminology and Glossary of Terms
The terminology used in the vitamin D field is confusing to many experts and nonexperts alike. A glossary of relevant terms is provided here to help the reader:
Vitamin D: Nutritional term coined in the 1920s to mean a substance that possesses the full antirachitic activity of the parent molecule vitamin D3. The term is now often employed as a short form for the class of compounds with the biologic activity of 1α,25-dihydroxyvitamin D, abbreviated as 1α,25-(OH)2D and also known as calcitriol. It is also occasionally used to mean a summation of both vitamin D2 and vitamin D3 forms (14), especially in the reporting of clinical assay results where serum 25-hydroxyvitamin D (25-OH-D) is used to mean the sum of 25-hydroxyvitamin D2 (25-OH-D2) and 25-hydroxyvitamin D3 (25-OH-D3) forms.
Vitamin D3: This natural derivative of 7-dehydrocholesterol is made in the skin (see Fig. 18.1 for structure).
Vitamin D2: Artificial or plant-derived form of vitamin D often used in food fortification, in daily dietary supplements, and in high-potency pharmaceutical preparations. It is metabolized to an active form, 1α,25-(OH)2D2, in a manner similar to the natural vitamin D3. Vitamins D2 and D3 are considered biologically equivalent in terms of their ability to cure rickets.
Renal 1α-hydroxylase: Proximal tubular form of the enzyme responsible for the final 1α-hydroxylation step of vitamin D activation, comprising three proteins, the key one being the cytochrome P-450 (CYP) protein CYP27B1. Renal 1α-hydroxylase is regulated by calcium (Ca2+) and phosphate (PO43-) ions through the hormones parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF-23).
Extrarenal 1α-hydroxylase: 1α-Hydroxylase, as evidenced by CYP27B1 protein, expressed in nonrenal tissues, especially where it locally produces 1α,25-(OH)2D3 that is believed to act in an autocrine or paracrine manner. It is regulated by cytokines and not by PTH and FGF-23.
Vitamin D receptor (VDR): Target cell protein that binds 1α,25-(OH)2D3, as well as specific sequences (vitamin D-responsive elements [VDREs]) in the genome to regulate gene expression of vitamin D-dependent genes at the transcriptional level. VDR does not bind any other metabolite with strong affinity, thus making it unlikely that vitamin D and 25-OH-D have any direct effect on gene expression under normal circumstances.
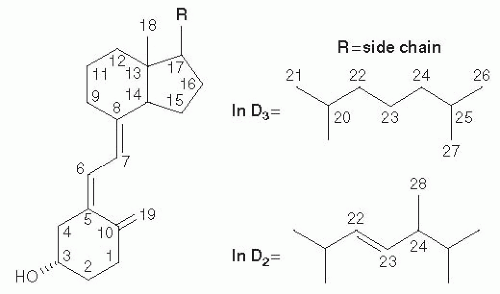
Fig. 18.1. Structure of vitamins D2 and D3. (Reprinted with permission from Makin HLJ, Jones G, Kaufmann M et al. Analysis of vitamins D, their metabolites and analogues. In: Makin HLJ, Gower DB, eds. Steroid Analysis. New York: Springer, 2010:967-1096.)
DIETARY SOURCES OF VITAMIN D
As already pointed out, vitamin D can be derived from either endogenous synthesis in the skin or from the diet. In the strictest use of the term, vitamin D is therefore not a vitamin, at least during the summer months. Consequently, it should be referred to as a prohormone. Dietary sources become critical during the winter months (above 43 degrees north between October and April) when the zenith angle of the sun is such that UVB light does not penetrate the atmosphere and synthesis of vitamin D3 in the skin is insignificant. Unfortunately, dietary sources of vitamin D are few, and most foodstuffs are devoid of vitamin D. The only significant sources of vitamin D (D2 or D3) are animal liver, fatty fish (e.g., salmon, halibut, cod), egg yolks, and fish oils. Unfortified cow’s milk is not a rich source. Because human milk is an extremely poor source of vitamin D, breast-fed infants require a vitamin D supplement. Most grains, lean meat, vegetables, and fruits are virtually devoid of measurable amounts of vitamin D. Although vitamin D2 can be derived from the plant sterol ergosterol, the likelihood that this provitamin D2 will be UV irradiated naturally seems low, even though some cultures sun-dry vegetables. The artificial irradiation of shitake mushrooms, a rich source of ergosterol, has increased the possibility of finding vitamin D2 in the diet.
Food fortification with either synthetic vitamin D2 or, later, vitamin D3 was pioneered and patented in the United States in the 1930s by Steenbock. His concept was to fortify staples such as breakfast cereals, milk, and margarine with vitamin D, initially in the form of irradiated ergosterol, to ensure delivery of this scarce nutrient and also to overcome the seasonal variability in potency found in natural sources (e.g., fish oils). This public health initiative to fortify certain foods with vitamin D eradicated rickets in the United States and virtually everywhere else in the world where it was introduced. In Quebec, Canada, which was the last province or state in North America to introduce vitamin D fortification, statistics showed a dramatic decline in the annual incidence of rickets cases at a downtown Montreal hospital (Ste Justine pour les Enfants) from 130 per 1000 to virtually 0 per 1000 over an 8-year span between 1968 and 1976; this period coincided with provincial legislation making it mandatory for dairies to fortify milk (15). A few countries resisted vitamin D fortification programs, not because they believed that they would be unsuccessful in the eradication of rickets but because of financial questions about whether government or industry would pay for the programs or out of concern about vitamin D intoxication in the neonatal period—fears that were proved largely unfounded.
RECOMMENDED DIETARY ALLOWANCES OF VITAMIN D
The dietary reference intakes (DRIs) were reviewed in a report published by a panel of the Institute of Medicine (IOM) at the National Academy of Sciences in Washington, DC (16). The chosen parameters for reporting optimal vitamin D intakes are currently adequate intake (AI) for the newborn to 1-year life stage and recommended dietary allowance (RDA) for all other life stages. Excessive intake is defined as tolerable upper intake level (UL) for all age groups; and the DRI values for the various life stage groups are provided in Table 18.1.
CURRENT UNDERSTANDING ABOUT VITAMIN D ACTIVATION AND INACTIVATION
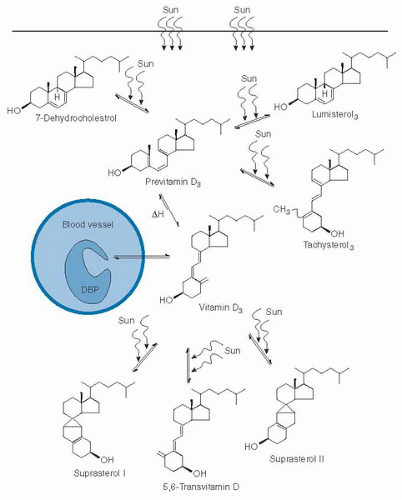
Vitamin D3 is synthesized from the sterol 7-dehydrocholesterol by a process involving UVB light in the wavelengths 290 to 315 nm (17) (Fig. 18.2). UV irradiation opens the 9,10-bond of the provitamin to give a previtamin D3 intermediate in the upper layers of the skin before it is isomerized nonenzymatically by heat to give vitamin D3 in the lower layers. Transport of vitamin D3 is carried by a specific plasma protein, vitamin D-binding protein (DBP), from skin to storage tissues or to the liver for the first step of activation. The D vitamins can also be derived from the diet, both as vitamin D3 and vitamin D2. Transport to storage depots or liver in the dietary case is on chylomicrons, although some evidence indicates that transfer from chylomicrons to DBP may also occur during transit. Vitamin D from skin or dietary sources does not circulate for long in the bloodstream, but instead is immediately taken up within hours by adipose tissue or liver for storage or activation (18).
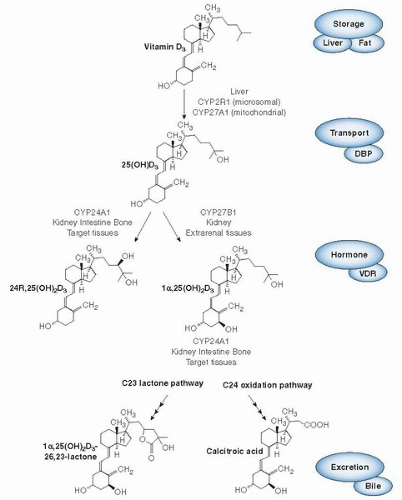
Ultimately, vitamin D3 undergoes its first step of activation (Fig. 18.3), namely, 25-hydroxylation in the liver. Over the years, some controversy has existed over whether 25-hydroxylation of vitamin D3 is carried out by one enzyme or two and whether this cytochrome P-450-based enzyme is found in mitochondrial or microsomal fractions of liver (18). Biochemical research has established that one human mitochondrial enzyme (CYP27A1) and several microsomal cytochrome P-450 enzymes (including CYP2R1, CYP3A4 and CYP2J3) are able to carry out the 25-hydroxylation of vitamin D2 or vitamin D3, or both (see Fig. 18.3) (reviewed in 19). The physiologic relevance of one of these enzymes, CYP2R1, is particularly pertinent because of a single report of a human mutation at Leu99Pro within the CYP2R1 gene in an individual with rickets (20); the enzyme is a 1α-OH-D2-25-hydroxylase with a high affinity for its vitamin D2 substrate (21). Investigations have provided a crystal structure of CYP2R1 with several of the known vitamin D substrates bound in the active site (22). Furthermore, a genome-wide association study of the genetic determinants of serum 25-hydroxyvitamin D concentrations (23) concluded that the chromosomal locus for CYP2R1 (11p15) showed the second strongest association of only a handful of sites in the whole genome; the others were DBP (or Gc), CYP24A1, and 7-dehydrocholesterol reductase (DHR7). Notably, variants of the other 25-hydroxylases, such as CYP27A1 or CYP3A4, were not identified as associated with serum 25-OH-D concentrations. This finding suggests that CYP2R1 is the most physiologically-relevant 25-hydroxylase.
TABLE 18.1 VITAMIN D DIETARY REFERENCE INTAKES BY LIFE STAGE (AMOUNT/DAY)
LIFE STAGE GROUP
AI
EAR
RDA
UL
Infants
0-6 mo
400 IU (10 µg)
—
—
1,000 (25 µg)
7-12 mo
400 IU (10 µg)
—
—
1,500 (38 µg)
Children
1-3 y
—
400 IU (10 µg)
600 IU (15 µg)
2,500 IU (63 µg)
4-8 y
—
400 IU (10 µg)
600 IU (15 µg)
3,000 IU (75 µg)
Males
9-13 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
14-18 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
19-30 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
31-50 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
51-70 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
>71 y
—
400 IU (10 µg)
800 IU (20 µg)
4,000 IU (100 µg)
Females
9-13 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
14-18 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
19-30 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
31-50 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
51-70 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
>71 y
—
400 IU (10 µg)
800 IU (20 µg)
4,000 IU (100 µg)
Pregnancy
14-18 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
19-31 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
31-50 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
Lactation
14-18 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
19-31 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
31-50 y
—
400 IU (10 µg)
600 IU (15 µg)
4,000 IU (100 µg)
AI, adequate intake; EAR, estimated average requirement; IU, international unit; RDA, recommended dietary allowance; UL, tolerable upper intake level.
Reproduced with permission from Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D. Washington, DC: National Academy Press, 2011.
Fig. 18.2. Photochemical events that lead to the production and regulation of vitamin D3 in the skin. DBP, vitamin D-binding protein; Sun, ultraviolet B rays as a component of sunshine. (Reprinted with permission from Holick MF. Photobiology of vitamin D. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D, 2nd ed. New York: Elsevier, 2005:37-46).
The product of the 25-hydroxylation step, 25-OH-D3, is the major circulating form of vitamin D3 and in humans is present in plasma at concentrations in the range 10 to 80 ng/mL (25 to 200 nmol/L) (24). The main reason for the extended plasma half-life of 25-OH-D3 is its strong affinity for DBP, and the DBP-XO mouse shows accelerated rates of clearance and low 25-OH-D3 levels (25). Serum levels of 25-OH-D3 therefore represent a measure of the vitamin D status of the animal in vivo.
The circulating metabolite, 25-OH-D3, is converted to the active form of vitamin D known as calcitriol or 1α,25-(OH)2D3. The second step of activation, 1α-hydroxylation, occurs primarily in the kidney (18). The synthesis of circulating 1α,25-(OH)2D3 in the normal, nonpregnant mammal appears to be the exclusive domain of the kidney. A specific mechanism appears to involve the cell surface receptors megalin and cubilin to provide uptake of the substrate, in the form of the 25-OH-D/DBP complex, by renal proximal cells (see Fig. 18.3) (26). Megalin knockout mice show reduced vitamin D metabolite levels and vitamin D deficiency. Additional evidence for renal synthesis of circulating calcitriol stems from clinical medicine. Patients with chronic kidney disease exhibit reduced 1α,25-(OH)2D3 levels and frank rickets or osteomalacia resulting from a deficiency of 1α,25-(OH)2D3 that is caused by lack of renal-1α-hydroxylase, a situation that can be reversed by 1α,25-(OH)2D3 hormone replacement therapy (27). The cytochrome P-450 enzyme, CYP27B1, representing the 1α-hydroxylase enzyme, was cloned virtually simultaneously from several species including rat, human, and mouse (28, 29, 30, 31). Investigators had known for some time that the kidney mitochondrial 1α-hydroxylase enzyme comprises three proteins—a cytochrome P-450, a ferredoxin, and a ferredoxin reductase for activity—and is strongly downregulated by 1α,25-(OH)2D3 and up-regulated by PTH as part of the calcium homeostatic loop (32). Investigators showed that a similar phosphate homeostatic loop also exists involving FGF-23, seemingly the long-postulated “phosphatonin” hormone that down-regulates CYP27B1 enzyme activity presumably at the transcriptional level (33). The promoter for the CYP27B1 gene appears to contain the necessary regulatory elements (cyclic AMP response elements [CREs] and negative VDREs) necessary to explain the observed physiologic regulations of PTH and 1α,25-(OH)2D3, respectively, at the transcriptional level. Whether additional elements exist to explain the action of FGF-23 through the klotho receptor remains to be addressed (33). Human CYP27B1 gene mutations result in vitamin D dependency rickets type 1 (VDDR-I) (34), a disease state first proposed, in 1973, to result from a genetic defect of the 1α-hydroxylase enzyme (35). Two independent groups generated the analogous mouse CYP27B1 knockout model by deletion of the cyp27B1 gene, and the 1α,25-(OH)2D3-deficient model revealed further insights into the regulation of the gene by different stimuli and subtleties in the roles of 1α,25-(OH)2D3 (36, 37).
Fig. 18.3. Metabolism of vitamin D3. Current knowledge of the key metabolites in vitamin D metabolism in conjunction with the enzymes (all cytochrome P-450s) involved in their production. The hormonal form, 1α,25-(OH)2D3, is the sole “active” form through a transcriptional mechanism involving target cell machinery, in particular the vitamin D receptor (VDR). The regulation of the enzyme system involves induction of CYP27B1 by parathyroid hormone and fibroblast growth factor 23 during a shortage of 1α,25-(OH)2D3 and induction of the CYP24A1 by the hormone itself during excess. Calcitroic acid is the biliary excretory product of 1α,25-(OH)2D3 generated by CYP24A1. A similar pathway exists for vitamin D2. DBP, vitamin D-binding protein.
The final piece of the vitamin D metabolic machinery has been elucidation of the catabolism of 25-OH-D3 and 1,25-(OH)2D3 in the body. This involves another cytochrome P-450 enzyme named CYP24A1, originally known as the 25-OH-D-24-hydroxylase. CYP24A1 inactivates both 25-OH-D3 and 1α,25-(OH)2D3 by subjecting these metabolites to 24-hydroxylation and thus giving rise initially to 24R,25-(OH)2D3 and 1α,24,25-(OH)3D3, respectively (38, 39) (see Fig. 18.3).
Currently, some speculation exists that 24-hydroxylated metabolites may play a unique biologic role in bone fracture repair, but most of the evidence favors the concept that 24-hydroxylation is primarily an inactivating step. The enzyme 24-hydroxylates both 25-OH-D3 and 1α,25-(OH)2D3, the latter with a 10-fold higher efficiency (40, 41). In the complete absence of DBP, however, this substrate discrimination is less evident. Because the circulating level of 25-OH-D3 is approximately 1000 times higher than that of 1α,25-(OH)2D3, the clearance of products of 25-OH-D3 by CYP24A1 (e.g., 24R,25-(OH)2D3) is readily evident in the bloodstream. The enzyme, particularly the renal form, appears to be expressed at high constitutive levels in the normal animal and may be involved in the inactivation and clearance of excess 25-OH-D3 from the circulation.
Conversely, the extrarenal 24-hydroxylase appears to be primarily involved in target cell destruction of 1α,25-(OH)2D3 (42). Indeed, CYP24A1 has now been shown to be expressed fairly ubiquitously, in particular wherever the VDR is expressed. CYP24A1 enzyme activity has been demonstrated in various cell lines representing specific vitamin D target organs (intestine, CaCo2 cells; osteosarcoma, UMR-106 cells; kidney, LLC-PK1 cells; keratinocyte, HPK1A and HPK1A-ras). Researchers have shown that 24-hydroxylation is the first step in the C-24 oxidation pathway, a five-step, vitamin D-inducible, ketoconazolesensitive pathway that changes the hydroxylated vitamins D molecules to water-soluble truncated products such as the biliary form, calcitroic acid (43, 44).
Although with human, rat, and mouse CYP24A1, the formation of calcitroic acid is the main pathway, other CYP24A1 isoforms, in particular the guinea pig and opossum analogs, predominantly carry out a 23-hydroxylation pathway to a 26,23-lactone product (45). The biologic value for the synthesis of the 26,23-lactone remains unknown, but the switch to the 23-hydroxylation pathway can be effected by a single Ala326Gly mutation in human CYP24A1 (45). In most biologic assays, the intermediates and truncated products of these 23- and 24-hydroxylation pathways possess lower or negligible biologic activity. Furthermore, many of these compounds have little or no affinity for DBP, thus making their survival in plasma tenuous at best. Polymerase chain reaction (PCR) studies of CYP24A1 led to the detection of CYP24A1 mRNA in a wide range of tissues, thereby corroborating the earlier studies reporting widespread 24-hydroxylase enzyme activity in most, if not all, calcitriol target cells.
Additional studies showed that mRNA transcripts for CYP24A1 are virtually undetectable in naive target cells, not exposed to 1α,25-(OH)2D3, but are dramatically induced by a VDR-mediated mechanism within hours of exposure to 1α,25-(OH)2D3 (46). In fact, the promoters of both human and rat CYP24A1 genes possess a double VDRE that has been shown to mediate the calcitriol-dependent induction of CYP24 enzyme in both species. It is therefore attractive to propose that not only is 24-hydroxylation an important step in the inactivation of excess 25-OH-D3 in the circulation but also it is involved in the inactivation of 1α,25-(OH)2D3 inside target cells. As such, one can hypothesize that C-24 oxidation is a target cell attenuation or desensitization process that constitutes a molecular switch to turn off calcitriol responses inside target cells (47).
Loss-of-function mutations of human CYP24A1 result in the condition known as idiopathic infantile hypercalcemia (IIH) (48) characterized by hypercalcemia, hypercalciuria, nephrolithiasis and nephrocalcinosis which supports a counterregulatory role for CYP24A1 in vitamin D metabolism. The CYP24A1-knockout mouse exhibits a similar hypercalcemic phenotype and in 50% of animals is severe enough to cause death at around weaning. Conversely, surviving animals have unexplained changes in bone morphology involving excess unmineralized osteoid that could suggest an alternative role for 24-hydroxylase in bone mineralization, although double knockouts lacking both CYP24A1 and VDR do not exhibit this bone phenotype (49). Surviving CYP24A1 null animals have been shown to possess a much reduced ability to clear a bolus dose of [1β-3H]1α,25-(OH)2D3 from their circulation as compared with normal, wild-type littermates (50). Thus, not much evidence exists for efficient non-CYP24A1-mediated backup excretory systems for calcitriol catabolism.
Calcitroic acid, the final water-soluble biliary product of 1α,25-(OH)2D3 catabolism, is probably not synthesized in liver because C-24 oxidation does not occur in hepatoma cells and presumably must therefore be transferred from target cells to liver by some plasma carrier. Although calcitroic acid has been found in various tissues in vivo (51), details of its transfer to bile have not been elucidated. Some emerging evidence indicates that high concentrations of vitamin D compounds can be metabolized by the general purpose liver cytochrome P-450 enzyme, CYP3A4, which is induced in intestine by calcitriol (52, 53, 54).
Only gold members can continue reading. Log In or Register to continue