Venous Thromboembolism
KEY CONCEPTS
![]() The risk of venous thromboembolism (VTE) is related to several easily identifiable factors, including age, major surgery (particularly orthopedic procedures of the lower extremities), previous VTE, trauma, malignancy, prolonged immobility or limb paralysis, and hypercoagulable states.
The risk of venous thromboembolism (VTE) is related to several easily identifiable factors, including age, major surgery (particularly orthopedic procedures of the lower extremities), previous VTE, trauma, malignancy, prolonged immobility or limb paralysis, and hypercoagulable states.
![]() The diagnosis of VTE should be confirmed by objective testing.
The diagnosis of VTE should be confirmed by objective testing.
![]() At the time of hospital admission, all patients should receive prophylaxis against VTE that corresponds to their level of risk. Prophylaxis should be continued throughout the period of risk.
At the time of hospital admission, all patients should receive prophylaxis against VTE that corresponds to their level of risk. Prophylaxis should be continued throughout the period of risk.
![]() In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., unfractionated heparin, low-molecular-weight heparin, fondaparinux, or rivaroxaban). If the patient is transitioned to warfarin therapy, the rapid-acting anticoagulant should be overlapped with warfarin for at least 5 days and until the patient’s international normalized ratio is greater than 2.
In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., unfractionated heparin, low-molecular-weight heparin, fondaparinux, or rivaroxaban). If the patient is transitioned to warfarin therapy, the rapid-acting anticoagulant should be overlapped with warfarin for at least 5 days and until the patient’s international normalized ratio is greater than 2.
![]() Most patients with an uncomplicated deep vein thrombosis (DVT), with or without pulmonary embolism (PE), can be safely treated as an outpatient.
Most patients with an uncomplicated deep vein thrombosis (DVT), with or without pulmonary embolism (PE), can be safely treated as an outpatient.
![]() Emerging anticoagulants such as rivaroxaban may mark a significant advancement in the treatment of VTE.
Emerging anticoagulants such as rivaroxaban may mark a significant advancement in the treatment of VTE.
![]() Anticoagulant therapies require meticulous and systematic monitoring as well as ongoing patient education.
Anticoagulant therapies require meticulous and systematic monitoring as well as ongoing patient education.
![]() Bleeding is the most common adverse effect associated with anticoagulant drugs. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, concurrent drug use, history of prior bleeding, prior history of stroke, renal or hepatic impairment, thrombocytopenia, recent surgery or trauma, and increasing age.
Bleeding is the most common adverse effect associated with anticoagulant drugs. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, concurrent drug use, history of prior bleeding, prior history of stroke, renal or hepatic impairment, thrombocytopenia, recent surgery or trauma, and increasing age.
![]() Anticoagulation therapies for VTE should be continued for a minimum of 3 months. The duration of anticoagulation therapy should be based on the patient’s risks of VTE recurrence and major bleeding, and preference regarding continued treatment.
Anticoagulation therapies for VTE should be continued for a minimum of 3 months. The duration of anticoagulation therapy should be based on the patient’s risks of VTE recurrence and major bleeding, and preference regarding continued treatment.
INTRODUCTION
Venous thromboembolism (VTE) is a potentially fatal disorder and a significant health problem in our aging society.1 Although young, otherwise healthy adults can have VTE, it most frequently occurs in patients who are hospitalized, undergo major surgery, are immobile for a lengthy period of time, or have a hypercoagulable disorder. VTE is manifested as deep vein thrombosis (DVT) and/or pulmonary embolism (PE) and results from clot formation within the venous circulation, most commonly the legs, but clots can also form in the arms, and in the mesenteric and cerebral veins (Fig. 9-1).1 DVT is rarely fatal, but death from PE can occur within minutes after symptom onset, before effective treatment can be given. Beyond the symptoms produced by the acute event, the complications of VTE, such as the postthrombotic syndrome and chronic thromboembolic pulmonary hypertension (CTPH), also cause substantial disability and suffering.1
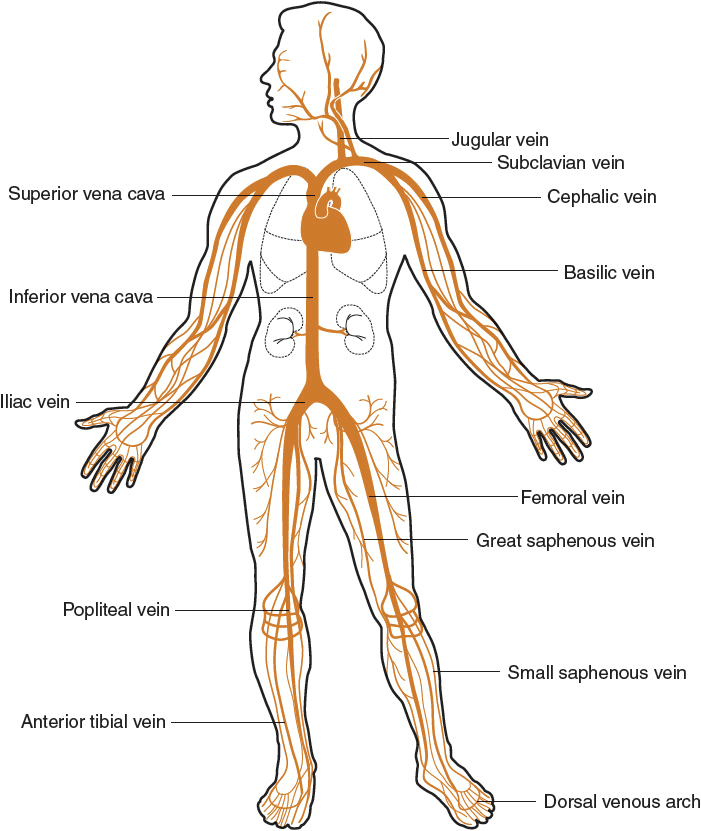
FIGURE 9-1 Venous circulation.
Anticoagulant drugs used in the prevention and treatment of VTE require precise dosing and meticulous monitoring.2,3 Systematic approaches to anticoagulant therapy management substantially reduce the risks, but bleeding remains an all too common and serious complication of administering anticoagulant drugs. Consequently, preventing VTE in at-risk patients is paramount to improving outcomes.4–6 When there is a suspicion of VTE, the rapid and accurate diagnosis of the disorder is critical to making appropriate treatment decisions.7 The optimal use of anticoagulant drugs requires an in-depth knowledge of their pharmacology and pharmacokinetic properties, and a comprehensive approach to patient management.2,3
EPIDEMIOLOGY
The true incidence of VTE in the general population is unknown because a substantial portion of patients have clinically silent disease. The annual incidence of symptomatic objectively diagnosed VTE is estimated at 2 to 3 per 1,000, increasing with age from 0.1 per 1,000 in adolescence to 8 per 1,000 in those over 80 years.8 The incidence of VTE in specific high-risk patient populations has been extensively studied.4–6 Patients sustaining multiple traumas or having orthopedic procedures involving the lower extremities are at particularly high risk as are those with a prior history of VTE and/or who have metastatic cancer. Likewise, the incidence of VTE after a myocardial infarction, stroke, and spinal cord injury is high. Several disorders of hypercoagulability have also been linked to a high lifetime incidence of VTE.4–6
ETIOLOGY
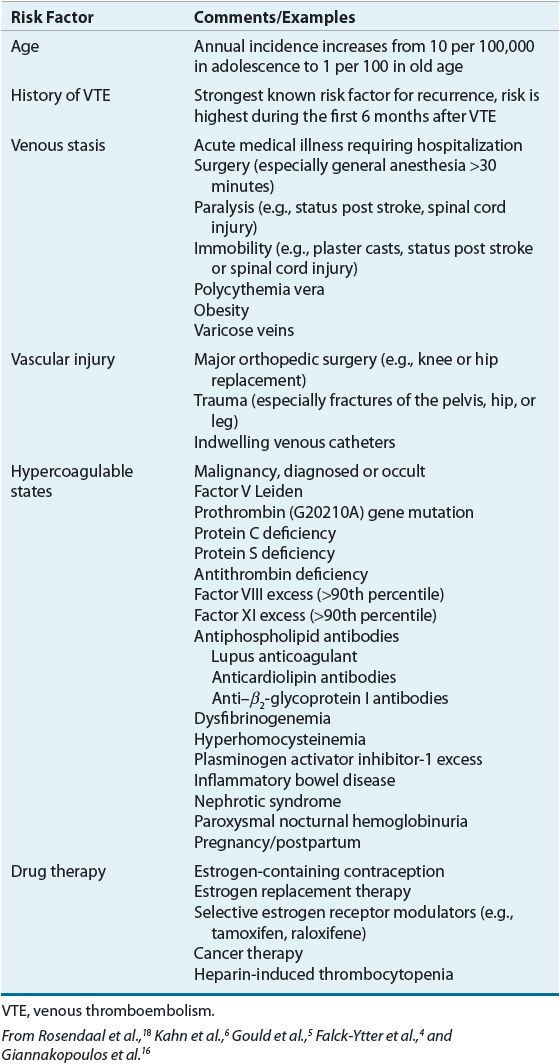
![]() A number of factors increase the risk of developing VTE (Table 9-1). The majority of these risk factors can be easily identified in clinical practice. Stasis of blood favors clotting in part through reduced clearance of activated clotting factors from sites of clot formation.9 The rate of blood flow in the venous circulation, particularly in the deep veins of the lower extremities, is relatively slow. Contraction of the calf and thigh muscles works with valves in the deep veins of the leg to facilitate the flow of blood back to the heart and lungs; thus, damage to the venous valves and periods of prolonged immobility result in venous stasis.8 Vessel obstruction, from either external compression or a thrombus, also promotes clot propagation. Reduced venous blood flow explains, at least in part, why numerous medical conditions and surgical procedures are associated with an increased risk of VTE (Table 9-1). Increased blood viscosity, seen in myeloproliferative disorders such as polycythemia vera, may also contribute to thrombus formation.8
A number of factors increase the risk of developing VTE (Table 9-1). The majority of these risk factors can be easily identified in clinical practice. Stasis of blood favors clotting in part through reduced clearance of activated clotting factors from sites of clot formation.9 The rate of blood flow in the venous circulation, particularly in the deep veins of the lower extremities, is relatively slow. Contraction of the calf and thigh muscles works with valves in the deep veins of the leg to facilitate the flow of blood back to the heart and lungs; thus, damage to the venous valves and periods of prolonged immobility result in venous stasis.8 Vessel obstruction, from either external compression or a thrombus, also promotes clot propagation. Reduced venous blood flow explains, at least in part, why numerous medical conditions and surgical procedures are associated with an increased risk of VTE (Table 9-1). Increased blood viscosity, seen in myeloproliferative disorders such as polycythemia vera, may also contribute to thrombus formation.8
TABLE 9-1 Risk Factors for Venous Thromboembolism
A growing list of inherited and acquired diseases has been linked to hypercoagulability (see Table 9-1). Activated protein C (aPC) resistance is the most common genetic disorder of hypercoagulability (prevalence rate of 2% to 7% in whites) accounting for many cases of DVT in unselected patients.10 Most aPC resistance results from a mutation on factor V that renders it resistant to degradation by aPC.10 This mutation is known as factor V Leiden, named after the city of Leiden, Holland, where the defect was first described. The prothrombin G20210A mutation is the second most frequent genetic risk factor, occurring in about 2% to 4% of whites and imparting about a threefold increased risk of VTE.10 This mutation increases circulating prothrombin, but the degree of VTE risk does not rise proportionally. Enhanced thrombin generation has been observed, but the mechanism whereby this disorder increases VTE risk is not completely understood.11 Although the rarity (present in <1% of the population) of inherited deficiencies of the natural anticoagulants protein C, protein S, and antithrombin precludes accurate quantification of their effect on the risk of initial VTE, many experts believe the lifetime risk is high.12 Conversely, excessively high concentrations of factors VIII, IX, and XI or fibrinogen also increase the risk of VTE.13 Given the prevalence of these inherited abnormalities in the general population, some patients have multiple genetic defects that have additive effects in terms of increasing the lifetime thrombotic risk.13
Acquired disorders of hypercoagulability may result from malignancy, the presence of antiphospholipid antibodies, and estrogen use.13 The strong link between cancer and thrombosis has long been recognized.14 Tumor cells secrete a number of procoagulant substances that activate the coagulation cascade. Furthermore, patients with cancer often have suppressed levels of endogenous anticoagulants (protein C, protein S, and antithrombin). It has been postulated that cancer cells use thrombotic mechanisms to recruit a blood supply (angiogenesis), metastasize, and create barriers against host defense mechanisms.15 Antiphospholipid antibodies are a heterogeneous group of antibodies targeting proteins that bind phospholipids.11 These include antibodies that prolong phospholipid-based clotting assays, known as lupus anticoagulants, as well as anticardiolipin and β2-glycoprotein (β2-gp) I antibodies. Antiphospholipid antibodies are found in up to 5% of normal healthy populations but are more common in patients with autoimmune disorders such as systemic lupus erythematosus and inflammatory bowel disease.16 Transient and low titers of these antibodies are common and do not present a risk for thrombosis. A diagnosis of antiphospholipid antibody syndrome requires the presence of moderate- or high-titer antibodies or lupus anticoagulants measured at least 12 weeks apart in a patient with a history of arterial or venous thrombosis or recurrent miscarriage.17 The precise mechanism by which antiphospholipid antibodies provoke thrombosis remains to be definitively determined. Contributing factors include complement activation, inhibition of proteins C and fibrinolysis, platelet activation, and increased tissue factor expression.16 Estrogen-containing contraception, estrogen replacement therapy, and the selective estrogen receptor modulators are all linked to venous thrombosis.18 Estrogens increase serum clotting factor concentrations and induce aPC resistance. Women with underlying hypercoagulability are at particularly high risk of developing venous thrombosis while taking estrogens.18
In many cases, VTE is the result of converging combinations of inherited and acquired thrombotic risk factors. Thus, many individuals with congenital hypercoagulable conditions experience a first VTE only after being placed in situations of high risk for thrombosis such as orthopedic surgery, immobilization, the use of estrogen-containing oral contraceptives, or pregnancy.19
PATHOPHYSIOLOGY
The arrest of bleeding following vascular injury is an amazingly complex process that is essential to life (Fig. 9-2). In the late 1800s, Dr. Rudolf Virchow, a German pathologist, recognized the role played by blood vessels, circulating elements in the blood, and the speed of blood flow in the regulation of clot formation.20 As described previously, alterations in any one of these elements, known today as Virchow’s triad, may lead to pathologic clot formation. Through the years various descriptive models of blood clot formation have been proposed and continuously refined based on new information and fresh thinking derived from increasingly sophisticated experimental methodology. Still many questions remain and a complete understanding of in vivo thrombus formation remains incomplete.21

FIGURE 9-2 Overview of hemostasis.
Hemostasis is the complex process responsible for maintaining the integrity of the pressurized circulatory system following damage to blood vessels.22 Hemostatic clots remain localized to the vessel wall and do not greatly impair blood flow in the vessel. In contrast, thrombotic clots such as those causing VTE result in impairment of blood flow and even complete occlusion of the vessel. Under normal circumstances, endothelial cells forming the intima of vessels maintain blood flow by physically separating extravascular collagen and tissue factor from platelets and clotting factors (namely, activated factor VII [VIIa]), respectively, thus preventing activation of hemostasis. Vascular injury allows key components of the coagulation process to seal the breach through the interaction of activated platelets recruited to the site of injury and the clotting factor cascade initiated by tissue factor and culminating in the formation of thrombin and ultimately fibrin clot (see Fig. 9-2).22 In contrast to physiologic hemostasis, pathologic venous thrombosis often occurs in the absence of gross vein wall disruption and may be triggered by circulating microparticles bearing tissue factor rather than the tissue factor expressed in the vessel wall. Venous clots often occur in areas of disturbed blood flow (e.g., valve cusps in the deep veins of the legs) and are mainly composed of fibrin with platelets and trapped red blood cells.21 Platelet thrombus and fibrin clot formation overlap temporally and occur nearly simultaneously,21 but are discussed separately below for simplicity.
Platelet Pathway
Platelets become actively involved in thrombus formation via two distinct pathways acting in parallel or separately. In one pathway, exposure of circulating blood to subendothelial collagen following vascular injury initiates platelet activation; in the other pathway platelets are activated by thrombin generated by tissue factor derived from the vessel wall or present in flowing blood.22 Various adhesion proteins (e.g., von Willebrand factor, fibrinogen) appear to play critical roles in platelet–vessel wall interactions.21 A platelet thrombus develops as activated platelets recruit unstimulated platelets, some of which are also activated while others remain loosely associated but do not undergo activation and may ultimately break away from the growing thrombus.22 Activation causes platelet α and dense granules to release their cargo of components such as adenosine diphosphate (ADP) and calcium ions that are critical for sustaining further thrombus formation into the environment surrounding the developing clot.22 Activated platelets accumulating in the thrombus also express P-selectin, an adhesion molecule that facilitates capture of blood-borne microparticles bearing tissue factor. Accumulation of tissue factor in the platelet thrombus is important as tissue factor is the primary initiator of fibrin clot formation via the coagulation cascade (Fig. 9-3).22
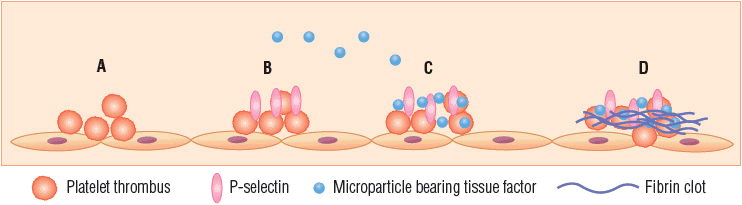
FIGURE 9-3 Model of pathologic thrombus formation: (A) activated platelets adhere to vascular endothelium; (B) activated platelets express P-selectin; (C) pathologic microparticles express active tissue factor and are present at a high concentration in the circulation—these microparticles accumulate, perhaps by binding to activated platelets expressing P-selectin; (D) tissue factor can lead to thrombin generation, and thrombin generation leads to platelet thrombus formation and fibrin generation. (Adapted from Furie and Furie.22)
Tissue Factor Pathway
The tissue factor pathway triggers coagulation by generating a small (picomolar) amount of thrombin. Before this small amount of thrombin is generated, the tissue factor pathway proceeds inefficiently through factor IX or X because factors VIII and V, the circulating procofactors required in the tenase and prothrombinase complexes, are not yet available in their most active cofactor form (Fig. 9-4). However, once formed, this small amount of thrombin converts factors VIII and V to their active cofactor forms, factors VIIIa and Va, respectively.22 The tenase and prothrombinase complexes now efficiently proceed to generate a large burst of thrombin. Even though the tissue factor pathway is rapidly downregulated, or inhibited, by the action of tissue factor pathway inhibitor, thrombin generation proceeds without replenishing active tissue factor.22
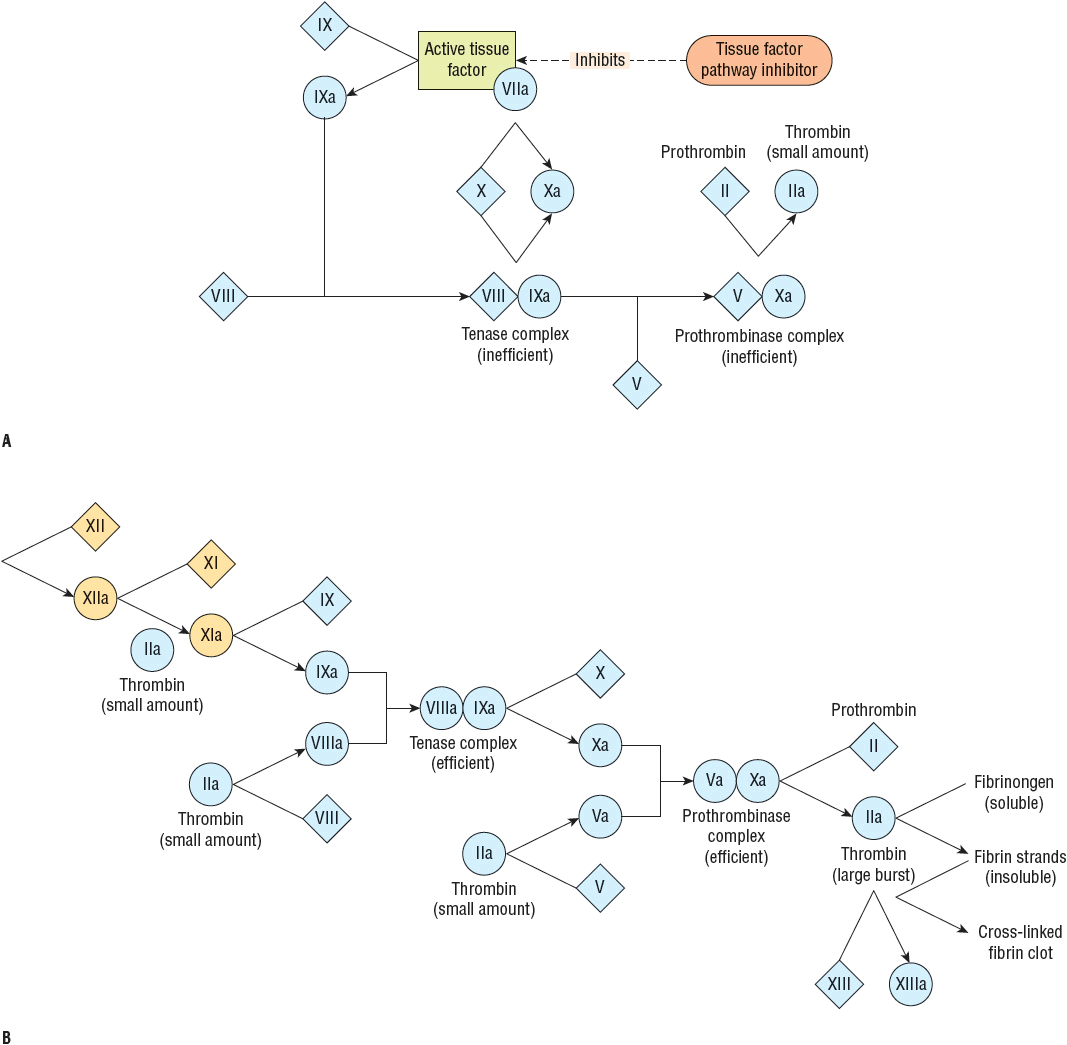
FIGURE 9-4 Coagulation cascade: (A) During the initiation phase the tissue factor–factor VIIa complex triggers blood coagulation by generating small amounts of thrombin. The tissue factor–factor VIIa complex activates factors IX and X. Factor IXa binds to factor VIII to form a tenase complex that inefficiently activates factor X to form factor Xa. Factor Xa, generated by the tissue factor–factor VIIa complex or the inefficient tenase complex, binds factor V on membrane surfaces to form a prothrombinase complex that inefficiently converts prothrombin to thrombin. The initiation phase is quickly downregulated by tissue factor pathway inhibitor. (B) During the amplification phase, the small amount of thrombin generated during initiation activates factors VIII and V, leading to a large burst of thrombin via highly efficient tenase and prothrombinase complexes. Thrombin converts fibrinogen to fibrin that forms strands that ultimately create an intricate web that traps red blood cells, platelets, and other cells to form a fibrin clot. Thrombin also activates factor XIII that cross-links fibrin to form a stabilized clot. (Adapted from Furie and Furie.22)
Fibrin Thrombus Formation
The final step in thrombus formation is the thrombin-mediated conversion of fibrinogen to form fibrin monomers. As fibrin monomers reach a critical concentration, they begin to precipitate and polymerize to form fibrin strands. Factor XIIIa, which is also activated by the action of thrombin, covalently bonds these strands to one another (Fig. 9-4) to form an extensive meshwork that surrounds and encases the aggregated platelet thrombus and red blood cells to form a stabilized clot that seals the site of vascular injury and prevents blood loss.23 Coagulation reactions are eventually terminated when this expanding meshwork of platelets and fibrin “paves over” the initiation site and additional activated factors are unable to diffuse through the overlying layer of clot.24
Endogenous Control of Thrombus Formation
Normally, a number of tempering mechanisms control coagulation (see Fig. 9-2).24 Without effective self-regulation, the coagulation cascade would continue unabated causing vascular occlusion at the site of injury. The intact endothelium adjacent to the damaged tissue actively secretes several antithrombotic substances.9 As its name implies, thrombomodulin modulates thrombin activity by converting protein C to its active form (aPC). When joined with its cofactor protein S, aPC enzymatically inactivates factors Va and VIIIa regulating the functionality of the prothrombinase and tenase complexes, respectively.9 The physiologic role of aPC is to prevent coagulation reactions from spreading to healthy, uninjured vessel walls. Antithrombin is a circulating protein that inhibits thrombin and factor Xa. Heparan sulfate, a heparin-like compound secreted by endothelial cells, exponentially accelerates antithrombin activity.9 By a similar mechanism, heparin cofactor II also inhibits thrombin. As described previously, tissue factor pathway inhibitor plays an important role by regulating the initiation of the coagulation cascade.22 When these self-regulatory mechanisms are intact, the formation of fibrin clot is limited to the zone of tissue injury. However, disruptions in the system, previously described hypercoagulable states, often result in pathologic thrombosis.19
Fibrinolysis
The fibrinolytic system is responsible for the dissolution of formed blood clots.25 An inactive proenzyme, plasminogen, is converted to an active enzyme, plasmin, that degrades the fibrin mesh into soluble end products collectively known as fibrin degradation products including D-dimer.25 The fibrinolytic system is also under the control of a series of stimulatory and inhibitory substances (see Fig. 9-2). Tissue plasminogen activator and urokinase plasminogen activator convert plasminogen to plasmin. Plasminogen activator inhibitor-1 inhibits the plasminogen activators, and α2-antiplasmin inhibits plasmin activity. Impaired activation of the fibrinolytic system has also been linked to hypercoagulability and thrombotic complications.25
Although a thrombus can form in any part of the venous circulation, the majority begin in the leg(s). Once formed, a venous thrombus may (a) remain asymptomatic, (b) spontaneously lyse, (c) obstruct the venous circulation, (d) propagate into more proximal veins, (e) embolize to the lungs resulting in PE, or (f) act in any combination of these ways.26 Thrombus isolated in veins of the calf is unlikely to embolize, but thrombus involving the popliteal and larger veins above it can break loose (embolize) and travel through the right side of the heart and lodge in the pulmonary artery or one of its branches, occluding blood flow to that part of the lung and impairing gas exchange. Without treatment, the affected portion of the lung becomes necrotic and oxygen delivery to other vital organs decreases, potentially resulting in fatal circulatory collapse.27
CLINICAL PRESENTATION AND DIAGNOSIS
Clinical Presentation
![]() The symptoms of DVT or PE (Tables 9-2 and 9-3) are nonspecific and objective tests are required to confirm or exclude the diagnosis.28 Patients with DVT frequently present with unilateral leg pain and swelling. Postthrombotic syndrome, a long-term complication of DVT caused by damage to the venous valves, may also result in chronic lower extremity swelling, pain, tenderness, skin discoloration, and, in the most severe cases, ulceration.7 PE typically presents with chest pain, shortness of breath, tachypnea, and tachycardia. PE is a life-threatening condition that may result in cardiopulmonary collapse.29
The symptoms of DVT or PE (Tables 9-2 and 9-3) are nonspecific and objective tests are required to confirm or exclude the diagnosis.28 Patients with DVT frequently present with unilateral leg pain and swelling. Postthrombotic syndrome, a long-term complication of DVT caused by damage to the venous valves, may also result in chronic lower extremity swelling, pain, tenderness, skin discoloration, and, in the most severe cases, ulceration.7 PE typically presents with chest pain, shortness of breath, tachypnea, and tachycardia. PE is a life-threatening condition that may result in cardiopulmonary collapse.29
TABLE 9-2 Clinical Presentation of Deep Vein Thrombosis

TABLE 9-3 Clinical Presentation of Pulmonary Embolism
Given that VTE can be debilitating or fatal, it is important to treat it quickly and aggressively.7 Conversely, because major bleeding induced by antithrombotic drugs can be equally harmful, it is important to avoid treatment when the diagnosis is not a reasonable certainty.28 Assessment of the patient’s status should focus on the search for risk factors in the patient’s medical history (see Table 9-1). Even in the presence of mild, seemingly inconsequential symptoms, VTE should be strongly suspected in those with multiple risk factors.
Diagnostic Studies
Radiographic contrast studies (venography and pulmonary angiography) are considered the gold standard in clinical trials because they are the most accurate and reliable method for diagnosing VTE.28 However, contrast studies are also expensive invasive procedures technically difficult to perform and evaluate. Severely ill patients are often unable to tolerate the procedure, and many develop hypotension and cardiac arrhythmias.28 Further, the contrast medium is nephrotoxic and irritating to the vessel wall and may paradoxically precipitate VTE.28 For these reasons, less invasive tests, such as compression ultrasound (CUS), computed tomography (CT) scans, and the ventilation–perfusion (V/Q) scan, are used in clinical practice for the initial evaluation of patients with suspected VTE.
D-Dimer
D-dimer is a degradation product of fibrin clot and levels of D-dimer are usually significantly elevated in patients with acute thrombosis.28 Although D-dimer is a very sensitive marker of clot formation, it is not sufficiently specific. A variety of conditions are associated with D-dimer elevations, including recent surgery or trauma, pregnancy, increasing age, and cancer; therefore, a positive D-dimer test is not conclusive evidence of VTE diagnosis. A wide variety of D-dimer assays of varying sensitivities for VTE are available. Enzyme-linked immunosorbent assays (ELISAs) and enzyme-linked immunofluorescence assays, along with the latex immunoturbidimetric assays, are generally termed “highly sensitive,” whereas the whole blood D-dimer assay is considered “moderately sensitive.”28 Appropriate use of D-dimer should include initial risk stratification via a validated clinical assessment model.28
Risk Stratification
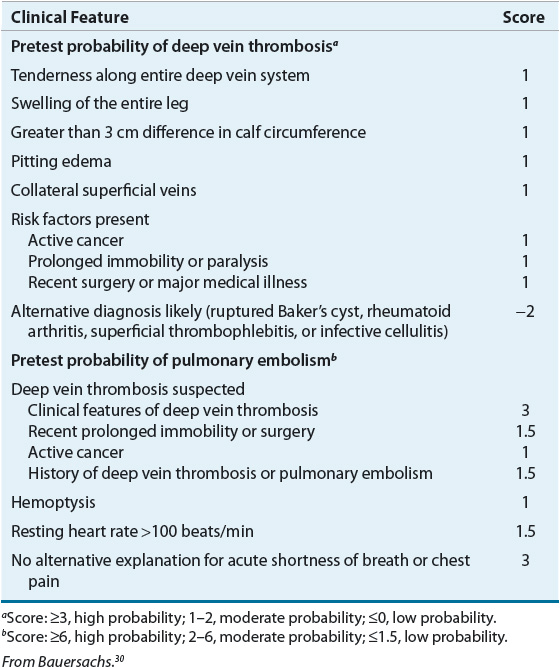
Clinical assessment significantly improves the diagnostic accuracy of noninvasive tests such as CUS, CT scanning, and D-dimer. Simple assessment checklists can be used to determine if a patient has a high, moderate, or low probability of DVT or PE (Table 9-4).30 Patients with a high pretest probability of VTE have a greater than 50% chance of having VTE, compared with only 5% of those with low pretest probability.31 Patients with a low pretest probability of VTE should receive testing with a moderate or highly sensitive D-dimer, if available.28 If the D-dimer result is normal, VTE is ruled out. Patients with a moderate pretest probability should receive either highly sensitive D-dimer or CUS. CUS may be performed proximally (i.e., above the knee) or throughout the entire leg. A normal full leg ultrasound or highly sensitive D-dimer rules out VTE. A normal proximal ultrasound should be repeated in 1 week or paired with D-dimer testing to rule out VTE. All patients with high pretest probability of VTE should receive either proximal or full leg CUS. A normal full leg ultrasound rules out VTE, whereas a normal proximal ultrasound requires additional testing with highly sensitive D-dimer, full leg ultrasound, or repeat proximal ultrasound surveillance in 1 week.28 Patients with CUS indicating proximal DVT should receive treatment, regardless of pretest probability. Evidence of calf vein DVT after full leg ultrasound may be managed by anticoagulation or further ultrasound surveillance to assess for propagation into the proximal deep veins of the leg.7,28
TABLE 9-4 Clinical Assessment Models for Deep Vein Thrombosis and Pulmonary Embolism
PREVENTION AND TREATMENT OF VENOUS THROMBOEMBOLISM
General Approach to the Prevention of Venous Thromboembolism
Given that VTE is potentially fatal and costly to treat, strategies to prevent DVT in at-risk populations positively impact patient outcomes.32 Relying on the early diagnosis and treatment of VTE is unacceptable because some patients will die before treatment can be initiated. Effective prophylaxis can reduce the risk of fatal PE in high-risk surgical and medical populations, whereas early ambulation is often sufficient for those at low risk of VTE.33 Educational programs and clinical decision support systems have been shown to improve the appropriate use of VTE prevention methods.34
![]() The most authoritative and well-recognized evidence-based guidelines for the prevention and treatment of VTE are the Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: Evidence-Based Clinical Practice Guidelines published by the American College of Chest Physicians (AT9). The AT9 attempt to balance concerns regarding the competing risks of symptomatic VTE and bleeding in their recommendations for thromboprophylaxis.33 Compared with previous guideline editions, the AT9 have critically evaluated the relevance of surrogate end points, such as venographically detected asymptomatic DVT, and based current recommendations mainly on the risk of symptomatic VTE (Table 9-5).33 An effective VTE prophylaxis program should identify and determine each patient’s level of risk for VTE and bleeding, and select and implement regimens that optimally balance these risks in a cost-effective manner. Several pharmacologic and mechanical methods are effective for preventing VTE, and these can be used alone or in combination.4–6 Mechanical methods improve venous blood flow, whereas drug therapy inhibits clotting factor activity or production. Despite ongoing efforts to minimize hospital-acquired VTE, up to one third of hospitalized patients at high risk of VTE without contraindications to anticoagulant therapy still do not receive appropriate prophylaxis.35
The most authoritative and well-recognized evidence-based guidelines for the prevention and treatment of VTE are the Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: Evidence-Based Clinical Practice Guidelines published by the American College of Chest Physicians (AT9). The AT9 attempt to balance concerns regarding the competing risks of symptomatic VTE and bleeding in their recommendations for thromboprophylaxis.33 Compared with previous guideline editions, the AT9 have critically evaluated the relevance of surrogate end points, such as venographically detected asymptomatic DVT, and based current recommendations mainly on the risk of symptomatic VTE (Table 9-5).33 An effective VTE prophylaxis program should identify and determine each patient’s level of risk for VTE and bleeding, and select and implement regimens that optimally balance these risks in a cost-effective manner. Several pharmacologic and mechanical methods are effective for preventing VTE, and these can be used alone or in combination.4–6 Mechanical methods improve venous blood flow, whereas drug therapy inhibits clotting factor activity or production. Despite ongoing efforts to minimize hospital-acquired VTE, up to one third of hospitalized patients at high risk of VTE without contraindications to anticoagulant therapy still do not receive appropriate prophylaxis.35
TABLE 9-5 Guidelines for the Prevention of Venous Thromboembolism

Clinical Controversy…
General Approach to the Treatment of Venous Thromboembolism
![]() Anticoagulation therapies remain the mainstay of treatment for VTE. DVT and PE are manifestations of the same disease process and are treated similarly.7 Before prescribing a full course of anticoagulation therapy for the treatment of VTE, it is imperative to establish an accurate diagnosis, thus preventing unnecessary risk of bleeding and expense to the patient (Fig. 9-5).7 Patients with high probability of VTE may need parenteral anticoagulation therapy while awaiting the results of diagnostic testing, whereas patients with intermediate probability may need parenteral anticoagulation only if diagnostic testing will be delayed more than 4 hours.7 The acute phase of VTE treatment (–7 days) requires rapidly acting anticoagulants such as unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), fondaparinux, or rivaroxaban to prevent thrombus extension and embolization. The early maintenance phase (7 days to 3 months) consists of continued therapeutic anticoagulation aimed at reducing the risk of long-term sequelae such as the postthrombotic syndrome and CTPH by allowing formed clot to be slowly dissolved by endogenous thrombolytic processes. Anticoagulation therapy extending beyond 3 months is aimed at long-term secondary prevention of recurrent VTE.1,7 Anticoagulation therapy is usually initiated with an injectable anticoagulant and then transitioned to warfarin maintenance therapy (Table 9-6 and Fig. 9-5). A simpler single-drug approach using orally administered rivaroxaban has now been shown to be noninferior to this standard approach for VTE treatment for appropriately selected patients.37,38
Anticoagulation therapies remain the mainstay of treatment for VTE. DVT and PE are manifestations of the same disease process and are treated similarly.7 Before prescribing a full course of anticoagulation therapy for the treatment of VTE, it is imperative to establish an accurate diagnosis, thus preventing unnecessary risk of bleeding and expense to the patient (Fig. 9-5).7 Patients with high probability of VTE may need parenteral anticoagulation therapy while awaiting the results of diagnostic testing, whereas patients with intermediate probability may need parenteral anticoagulation only if diagnostic testing will be delayed more than 4 hours.7 The acute phase of VTE treatment (–7 days) requires rapidly acting anticoagulants such as unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), fondaparinux, or rivaroxaban to prevent thrombus extension and embolization. The early maintenance phase (7 days to 3 months) consists of continued therapeutic anticoagulation aimed at reducing the risk of long-term sequelae such as the postthrombotic syndrome and CTPH by allowing formed clot to be slowly dissolved by endogenous thrombolytic processes. Anticoagulation therapy extending beyond 3 months is aimed at long-term secondary prevention of recurrent VTE.1,7 Anticoagulation therapy is usually initiated with an injectable anticoagulant and then transitioned to warfarin maintenance therapy (Table 9-6 and Fig. 9-5). A simpler single-drug approach using orally administered rivaroxaban has now been shown to be noninferior to this standard approach for VTE treatment for appropriately selected patients.37,38
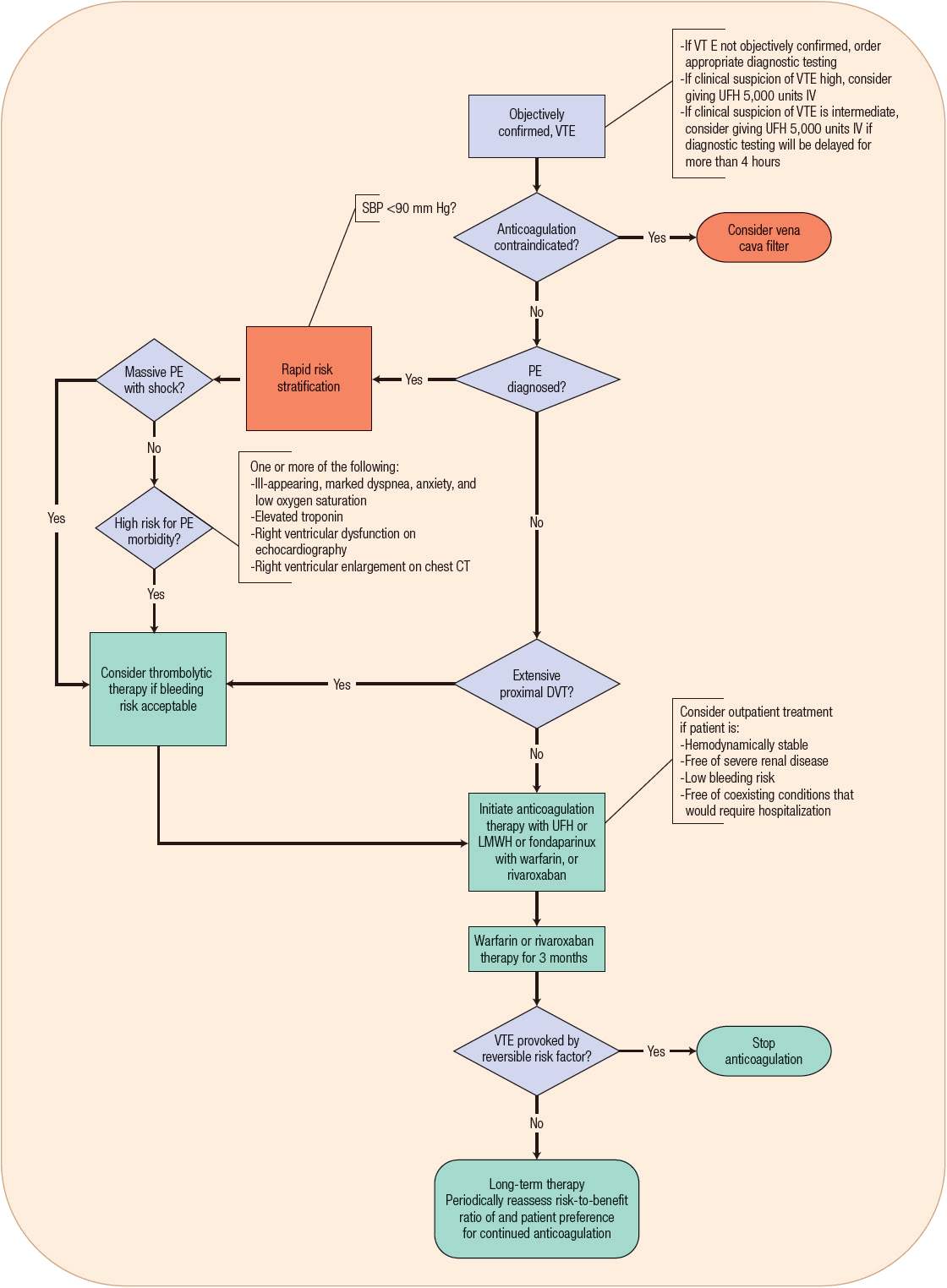
FIGURE 9-5 Treatment of venous thromboembolism (VTE). (DVT, deep vein thrombosis; LMWH, low-molecular-weight heparin; PE, pulmonary embolism; SBP, systolic blood pressure; UFH, unfractionated heparin.)
TABLE 9-6 Guidelines for the Treatment of Venous Thromboembolism

Determining the optimal duration of anticoagulation involves weighing the risk of recurrent VTE against the risk of bleeding associated with anticoagulation therapy and determining patient preference regarding treatment duration (see Evaluation of Therapeutic Outcomes below). When bleeding risk outweighs VTE recurrence risk or if the informed patient’s preference is to stop treatment, anticoagulation therapy should be discontinued.7 In life- or limb-threatening circumstances, elimination of the obstructing thrombus may be warranted and the use of thrombolysis or thrombectomy can be considered.7 Insertion of a removable filter in the inferior vena cava (IVC) is also an option in those with contraindications to anticoagulation therapy or in whom anticoagulant therapy has failed.7
![]() Once the diagnosis of VTE has been objectively confirmed (see Clinical Presentation and Diagnosis above), anticoagulant therapy with UFH, LMWH, fondaparinux, or rivaroxaban should be instituted as soon as possible. Evidence indicates that currently available injectable anticoagulants can be administered in the outpatient setting in most patients with DVT and in carefully selected hemodynamically stable patients with PE (see Table 9-6).1 The decision to initiate therapy on an outpatient basis should be based on institutional resources and patient-specific variables (Table 9-7).39
Once the diagnosis of VTE has been objectively confirmed (see Clinical Presentation and Diagnosis above), anticoagulant therapy with UFH, LMWH, fondaparinux, or rivaroxaban should be instituted as soon as possible. Evidence indicates that currently available injectable anticoagulants can be administered in the outpatient setting in most patients with DVT and in carefully selected hemodynamically stable patients with PE (see Table 9-6).1 The decision to initiate therapy on an outpatient basis should be based on institutional resources and patient-specific variables (Table 9-7).39
TABLE 9-7 Outpatient Treatment Protocol for Deep Venous Thrombosis

NONPHARMACOLOGIC THERAPY
Mechanical Prevention and Treatment Strategies
Graduated compression stockings and intermittent pneumatic compression (IPC) devices improve venous blood flow and reduce the risk of VTE. These methods do not increase the risk of bleeding, which makes them attractive for postoperative VTE prophylaxis. However, they are not risk free as skin breakdown and ulceration can occur.6
Graduated compression stockings have failed to reliably demonstrate a reduction in VTE in medically ill patients.6 However, they reduce the incidence of VTE (including asymptomatic and distal DVT) by approximately 65% when used after orthopedic surgery, cardiac surgery, gynecologic surgery, or neurosurgery.5 Compression stockings work by increasing the velocity of venous blood flow through the application of graded pressure, with the greatest amount applied at the ankle. This treatment option is inexpensive and safe, and an excellent choice when pharmacologic intervention is either contraindicated or difficult to adequately monitor.6 When combined with pharmacologic interventions, graduated compression stockings have an additive effect. However, they can be uncomfortable and some patients are unable to wear them because of the size or shape of their legs and adverse skin reactions.6
IPC devices utilize sequential inflation of a series of cuffs wrapped around the patient’s legs to increase the velocity of blood flow.6 The cuffs use graded pressure and inflate in 1- to 2-minute continuous cycles from the ankles to the thighs. IPC reduces the risk of VTE by more than 60% following general surgery, neurosurgery, and orthopedic surgery.5 Although IPC is well tolerated and safe to use in patients who have contraindications to pharmacologic therapies, it does have drawbacks. There is some theoretical concern that external compression may dislodge a previously formed clot, IPC is more expensive and cumbersome to use than graduated compression stockings, and some patients have difficulty sleeping and getting in and out of bed while wearing the devices.4 For optimal effectiveness, IPCs should be worn at least 18 hours/day. Battery-operated units facilitate mobility and monitoring. Like graduated compression stockings, IPCs can be used in combination with pharmacologic strategies.4
IVC filters can provide short-term protection against PE in very-high-risk patients by blocking embolization of thrombus formed below the filter.5 Percutaneous insertion of an IVC filter is a minimally invasive procedure performed using fluoroscopic imaging to verify placement. Despite the widespread use of IVC filters, only limited data support effectiveness and long-term safety in the setting of VTE prevention. Nonrandomized data suggest IVC filters reduce the short-term risk of PE but are associated with complications including DVT, filter migration, IVC occlusion, and insertion site thrombosis.5 IVC filters should be reserved for patients at highest risk for VTE in whom other prophylactic strategies cannot be used.
IVC filters also have a limited role in the management of acute VTE when anticoagulants are ineffective or unsafe, including in (a) patients with an absolute contraindication to anticoagulation therapy because of active bleeding or anticipated bleeding from a preexisting lesion; (b) patients with massive PE who survive but in whom recurrent embolism might be fatal; (c) patients who have recurrent VTE despite adequate anticoagulation therapy; and (d) demonstration of large free-floating clot loosely attached to the wall of the IVC.40 IVC filters reduce the risk of PE in the short term, but also appear to increase the long-term risk for recurrent DVT presumably as a consequence of the accumulation of thrombus on the filter, which may partially occlude the vena cava, resulting in venous stasis.40 Retrievable filters that can be removed after the period of greatest risk for PE or after a transient bleeding risk has resolved have been developed and suggested for use in patients with transient contraindications to anticoagulation therapy. In reality most (65% in one report) “retrievable” filters are not removed.40 When an IVC filter is inserted as an alternative to anticoagulation, the AT9 suggest a conventional course of anticoagulant therapy once the risk of bleeding resolves.7
Ancillary Therapy
In addition to anticoagulant therapy for patients with proximal DVT, wearing graduated compression stockings can reduce the risk of developing the postthrombotic syndrome by 50%.7 To be effective, graduated compression stockings must fit properly and provide adequate pressure at the ankle (30 to 40 mm Hg; 4 to 5.3 kPa).7 Discomfort and unflattering appearance are barriers to adherence with compression stockings, particularly during the hot summer months. However, consistent daily use should be encouraged for at least 2 years after DVT or longer if symptoms of postthrombotic syndrome persist.7
Strict bedrest was traditionally recommended following acute DVT based on the assumption that leg movement would dislodge the clot, resulting in PE. However, ambulation in conjunction with graduated compression stockings results in faster reduction in pain and swelling with no apparent increase in the rate of embolization. Early ambulation and continued high activity level also reduce the likelihood of postthrombotic syndrome.7 Patients should be encouraged to ambulate as much as their symptoms permit. If pain and swelling increase with ambulation, the patient should be instructed to lie down and elevate the affected leg until symptoms subside.
PHARMACOLOGIC STRATEGIES FOR VTE PREVENTION
Pharmacologic options for preventing VTE have been extensively evaluated in randomized clinical trials (see Table 9-5). Appropriately selected drug therapies can significantly reduce the incidence of VTE following hip and knee replacement, hip fracture repair, general surgery, myocardial infarction, ischemic stroke, and in appropriately selected hospitalized medical patients.4–6 The optimal agent and dose to use for VTE prevention must be based on the patient’s level of risk for thrombosis and bleeding complications, as well as cost and availability.
Medical Patients
Several risk assessment models have been developed to identify hospitalized and critically ill patients at high risk of VTE who would realize the greatest benefit from thromboprophylaxis. The Padua Prediction Score is a prospectively validated VTE risk assessment tool for hospitalized medical patients.6 For this tool, 3 points each are assigned for active cancer, previous VTE, reduced mobility, and thrombophilia; 2 points are assigned for trauma and/or surgery within the last month; and 1 point each is assigned for age ≥70 years, heart and/or respiratory failure, acute myocardial infarction or ischemic stroke, acute infection and/or rheumatologic disorder, body mass index ≥30 kg/m2, or ongoing hormonal treatment. Among high-risk patients (score ≥4 points) not receiving prophylaxis, VTE occurred in 11% within 90 days compared with just 0.3% of low-risk patients.6
Recommendations for preventing VTE during medical illness are summarized in Table 9-5. Compared with placebo, low-dose unfractionated heparin (LDUH), LMWH, and fondaparinux all reduce the risk of symptomatic VTE and fatal PE among high-risk medical patients.6 Hospitalized and acutely ill medical patients at high risk of VTE and low risk of bleeding should receive pharmacologic prophylaxis with LDUH, LMWH, or fondaparinux for the duration of hospitalization or until fully ambulatory. Routine use of pharmacologic prophylaxis is not warranted in low-VTE-risk populations. In medical patients at high risk of bleeding who also require VTE prophylaxis, mechanical prophylaxis may be preferred over anticoagulation therapy.6 The AT9 guidelines suggest that mechanical prophylaxis may be preferred in patients with any of the following bleeding risk factors: active gastric or duodenal ulcer, history of bleeding within 90 days, or platelet count <50 × 109/L.6 Mechanical prophylaxis should also be considered if more than one of the following are present: Age 85 years or more, hepatic failure, renal failure (creatinine clearance <30 mL/min), admission to intensive care or cardiac care units, central venous catheter, rheumatic disease, active cancer, or male sex.6
Nonorthopedic Surgery Patients
Patient risk for VTE after general surgery can be estimated using the Caprini score.5 This extensive risk assessment tool awards 1, 2, 3, or 5 points to patient-specific risk factors (e.g., age, BMI, VTE history) and procedure-related risk factors including minor or major surgery, laparoscopic or open procedures, and elective arthroplasty. Once all risk factor points are summed, VTE risk is categorized as very low (0 to 1 point), low (2 points), moderate (3 to 4 points), or high (≥5 points).5 Estimating bleeding risk associated with surgery is challenging due to wide variety of surgery types, the effect of surgical technique, and the lack of a validated clinical predication rule. Table 9-5 summarizes the AT9 recommendation for preventing VTE following nonorthopedic surgery. In general, patients at high risk of VTE but at low risk of bleeding should receive prophylaxis with LDUH or LMWH in addition to graduated compression stockings or IPC. Patients at high risk of bleeding should receive IPC if the risk of VTE is moderate or high.5
Orthopedic Surgery
Total joint arthroplasty is associated with a very high risk of postoperative VTE. Recommended pharmacologic agents for the prevention of VTE following joint replacement surgery include aspirin, adjusted-dose warfarin, UFH, LMWH, fondaparinux, dabigatran, apixaban, and rivaroxaban for a minimum duration of 10 days postsurgery.4 Head-to-head trials of these agents fail to reliably demonstrate differences in clinically relevant outcomes such as symptomatic VTE, fatal PE, major hemorrhage, and surgical site complications.4 The endorsement of aspirin for VTE prevention in the AT9 is a major departure from previous guidelines that actually recommended against aspirin use in this setting.4,41 The change in recommendation reflects recognition by AT9 panelists that previous recommendations did not reflect actual clinical practice due to overreliance on surrogate end points such as asymptomatic DVT detected via screening venography.33
The AT9 cite extensive clinical experience and similar or superior properties compared with other pharmacologic options as reasons for suggesting LMWH as preferred over other agents after total joint arthroplasty despite a lack of convincing evidence of superior efficacy or safety compared with older, less costly agents.4 The risk of bleeding associated with LMWH use following orthopedic surgery relates closely to the timing of thromboprophylaxis initiation. Administration of LMWH within 2 hours preoperatively or postoperatively increases the risk of bleeding up to fivefold compared with starting 12 hours after surgery.4
Warfarin remains among the most commonly prescribed agents for VTE prevention after total joint arthroplasty offering some advantages over LMWH, including low acquisition cost and oral administration.42 Warfarin’s delayed onset of anticoagulant effect confers both a potential advantage (reduced immediate risk of postoperative bleeding) and a disadvantage (increased risk of early VTE). Previous American College of Chest Physician’s guidelines recommended a goal international normalized ratio (INR) range of 2 to 3 following orthopedic surgery while American Academy of Orthopaedic Surgery (AAOS) guidelines recommended an INR target of 2 or “one which appropriately balances the risk of PE and bleeding.”41,43 The AT9 now simply recommend “dose-adjusted warfarin” without specific guidance on target INR, and the AAOS guidelines also no longer recommend a specific INR target.4,43 Many orthopedic surgeons prefer low-intensity warfarin (e.g., INR 1.5 to 2.5) due to perceived lower risk of postoperative bleeding.42 Regardless of INR target, warfarin use following orthopedic surgery requires a well-coordinated monitoring system and timely INR testing.44 Arranging INR testing for patients following joint replacement surgery can be challenging due to limited patient mobility often requiring arrangement of home phlebotomy or use of point-of-care INR monitoring devices that erodes the cost advantage of warfarin.
Rivaroxaban, apixaban, and dabigatran offer the convenience of oral administration and fixed dosing without the need for routine coagulation testing. Clinical trials of these agents have demonstrated safety and efficacy similar to enoxaparin after total joint replacement, but they have not been studied after hip fracture surgery.4 The AT9 express a preference for apixaban or dabigatran (alternatively warfarin or rivaroxaban) in patients unwilling to use injections of LMWH, but neither dabigatran nor apixaban is FDA approved for VTE prevention after total joint arthroplasty.4 Postmarketing studies are needed to provide real-world context in a variety of patient populations to the favorable results of initial clinical trials for these new anticoagulants.
Duration of Therapy
The optimal duration of VTE prophylaxis following surgery is not well established.41 Prophylaxis should be given throughout the period of increased VTE risk. For general surgical procedures and medical conditions, once the patient is able to ambulate regularly and other risk factors are no longer present, prophylaxis can be discontinued.5,6 Because of the relatively high incidence of VTE in the first month following hospital discharge among patients who have undergone a lower extremity orthopedic procedure, extended prophylaxis following hospital discharge appears to be beneficial.4 Most clinical trials support the use of antithrombotic therapy for 21 to 35 days following total hip replacement and hip fracture repair surgeries.4
Pharmacoeconomic Considerations in VTE Prevention
The acquisition costs of graduated compression stockings, UFH, and warfarin are considerably less than those of LMWH, fondaparinux, rivaroxaban, and dabigatran. However, the drug acquisition cost is relatively small when compared with the overall cost of care.32 Economic analyses must take into account therapeutic efficacy, duration of use, complications, and monitoring costs.45
The determination of the cost-effectiveness of VTE prophylaxis is based on the premise that a reduction in future VTE events will reduce overall healthcare costs.32 The cost of providing VTE prophylaxis for 1,000 patients declines as the incidence of VTE in a given population increases. Compared with no prophylaxis, prophylaxis with UFH or LMWH in immobilized medically ill patients appears to be cost-effective, with incremental costs ranging from $65 to $2,534 per quality-adjusted life-year.6 Studies of nonorthopedic surgical populations suggest that use of graduated compression stockings, IPCs, LMWH, and LDUH is more cost-effective than no prophylaxis.5 Pharmacologic prophylaxis in high-risk surgical and orthopedic populations is likely to be cost-effective compared with no prophylaxis given the high risk for postoperative VTE. In this population a systematic review showed that fondaparinux was more cost-effective than LMWH, and that warfarin and LMWH were similarly cost-effective.46 The availability of generic enoxaparin may alter these findings. Extended prophylaxis is cost-effective after total hip arthroplasty, but the results of cost analysis after total knee arthroplasty are inconclusive.46
PHARMACOLOGIC STRATEGIES FOR VTE TREATMENT
Unfractionated Heparin
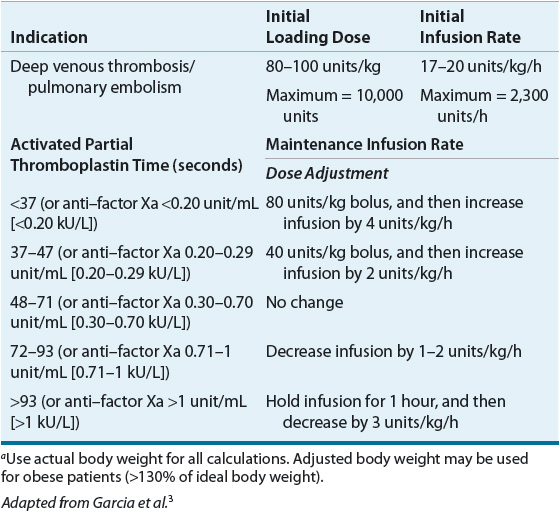
The parenteral administration of UFH is preferred for acute VTE treatment in patients with severe renal insufficiency (creatinine clearance <30 mL/min [<0.5 mL/s]).7 UFH may be administered subcutaneously (SC) with or without coagulation monitoring or by continuous IV infusion (see Table 9-8). Because the anticoagulant response to UFH is highly variable, it is standard practice to adjust the dose based on the results of coagulation tests. When UFH is administered by IV infusion, the activated partial thromboplastin time (aPTT) is generally used to monitor the anticoagulant effect, although aPTT response varies between laboratories using different reagents and instruments and methods for improving interlaboratory agreement (e.g., standardization of aPTT ratios by reference to anti-Xa levels) have had limited success.3 For these reasons, the therapeutic aPTT range at each institution should be adapted to the responsiveness of the reagent and instrument used.3 Either weight-based dosing (see Table 9-8) or fixed dosing (e.g., 5,000 unit bolus followed by 1,000 units/h continuous infusion) of UFH produces similar clinical outcomes.44 However, failure to give a sufficient IV UFH dose has been shown to increase the risk of VTE recurrence not only during initial treatment but also during long-term therapy.3 IV UFH requires hospitalization with frequent aPTT monitoring and dose adjustment. Inpatient anticoagulation management services have been shown to improve patient care by increasing the proportion of aPTT values in the therapeutic range, reducing the length of hospital stay, and lowering total hospital costs when compared with usual care.47 However, some patients still fail to achieve an adequate response to UFH therapy.3 Consequently, traditional IV UFH in the acute treatment of VTE has largely been replaced by LMWH or fondaparinux. However, as clearance of LMWH, fondaparinux, and the new oral anticoagulants is dependent in some degree on renal function, UFH will continue to have a role for acute VTE treatment in patients with creatinine clearance <30 mL/min (<0.5 mL/s).7
TABLE 9-8 Weight-Baseda Dosing for Unfractionated Heparin Administered by Continuous IV Infusion