Fig. 9.1
Argon laser with 488 nm wavelength. Photon release of light with characteristic wavelength. FITC 530 nm; PE 578 nm; PE-Cy5 670 nm
Clinical Application
The continued interest in stem cell transplantation research and growth in its clinical applicability has grown tremendously in the last 20 years. Stem cells, by their nature, are defined as cells that are quiescent and multipotent and have the capacity for self-renewal and ultimate differentiation [16]. The success of using bone marrow transplantation to treat blood-borne malignancies has led to the use of using stem cell for solid organ transplantation with improved outcomes [17]. The adverse effects associated with chronic immunosuppression, rejection, and the overall increase in need for solid organ availability have led to the increased interest in transplantation and regenerative medicine [16].
In addition to the possible benefits of stem cell transplantation in clinical medicine, there must also be an awareness to the potential dangers. Among these is the possibility of graft vs. host disease (GVHD) in the stem cell transplant recipient. Stem cells, by their multipotent nature, can be potentially tumorigenic or infectious [16]. Thus, the clinician must weigh the benefit to risk ratio when treating a patient with stem cell transplantation therapy. Flow cytometry has a role in assisting the clinician’s armamentarium when deciding the best treatment course for a patient. Flow cytometry enhances the ability to understand the molecular and immunologic mechanisms involved in stem cell differentiation and maturation.
Flow cytometry is able to analyze large populations and disparate cell types via the measurement of cellular DNA content [14]. The fluorescence emitted is directly proportional to the amount of DNA in the nucleus. In addition, cell-surface proteins on various cell lines have been studied in great detail as well. Fluorescent-tagged antibodies to either extracellular or intracellular proteins allow the analysis of erythrocytes, leukocytes, and platelets.
One such cell-surface protein is the cluster of differentiation 4 (CD4), a glycoprotein found on T lymphocytes, among other immune cells. With the ability to target CD4+ T cells directly and quantitatively, one is able to study the hematologic workup of various leukemias and lymphomas. To distinguish between the various subtypes of leukemias and lymphomas via flow cytometry, analysis of their antigens has better enabled the physician to diagnose, treat, and provide prognosis to patients. The direct correlation of fluorescence and disease is not only limited to blood dyscrasias. Flow cytometric analysis of specific immunologic cell subsets in an allogeneic stem cell graft can serve as a predictor for graft survival [18, 19]. Work performed by Waller et al. and Reddy et al. demonstrated that immune reconstitution following allogeneic bone marrow transplantation can be quantified by flow cytometric analysis [19, 20].
The ability to identify specific antigens on cell surfaces via flow cytometry has improved the physician’s ability to detect, isolate, and quantify cells of interest in a much improved and less cumbersome fashion. This ability has played a major role in the field of transplantation. In 1983, work by Garovoy et al. demonstrated the importance of human leukocyte antigen (HLA) typing [21]. They demonstrated in the kidney transplant that donor-directed HLA antibodies (Abs) detected by flow cytometric analysis could potentially induce early rejection and loss. Further studies confirmed that identifying HLA Abs that were directed against mismatched HLA antigens (Ags) would lead to failure of engraftment not only in kidney transplants but in other solid organs as well [22–25].
The ability to identify T lymphocytes and B lymphocytes based on cell-surface protein expression also led to the ability to identify and sort stem cells. Civin et al. in 1984 demonstrated that CD34 was a marker for identifying hematopoietic stem cells [26]. This led to interest in using flow cytometry as a method for identifying and sorting CD34+ cells that were to be used in transplantation.
Analyzing the Data
In principle, all flow cytometers analyze particles in the same manner. However, if the cytometer is not configured correctly, the data gathered could be rendered meaningless [13]. An inexperienced user may not even realize that they have not configured the machine incorrectly and may be acquiring non-useful data inadvertently. Thus, it is imperative that the instrument is configured correctly.
Initially, the user must calibrate several control samples that are imperative to a proper configuration of the flow cytometer. These control samples serve as reference points for the acquired data in the experimental samples [13]. There are three basic types of control samples: negative, positive, and compensation control samples. Negative control samples adjust the instrument parameters so that all data appear on scale. Positive control samples confirm that the fluorochromes (conjugated antibodies) are able to recognize the antigen. And compensation control samples are used for multicolor to adjust for overlap [13].
The data acquired is most commonly displayed in a histogram. Histograms can be configured to display any parameter chosen. They are excellent because they permit the researcher to visualize the distribution of single measured parameter [13]. By overlaying multiple histogram samples on one parameter plot, one is able to compare fluorescence or other measured parameters. By setting statistical markers, data obtained can be measured quantitatively.
Another important feature of flow cytometry analysis is the possibility of gating [1]. By selecting a cell subset within a group of cells, gating allows for further analysis. Gating is performed by enclosing a cluster of cells in a data plot that exhibits similar characteristics based on light scatter. Fluorescence can then be further analyzed in the gated region based on previously defined parameters (e.g., forward scatter, side scatter, green fluorescence vs. red). Gating is not only an inclusion method, i.e., analyzing subset of a cell population; it can also be used as an exclusion method—small cellular debris or large aggregates [13]. The combination of fluorescence and light-scattering properties are invaluable in analyzing data. Additional information, such as increased background fluorescence from nonspecific binding of conjugated antibodies or entrapment of fluorescently labeled antibodies in dead, dying cells, or debris, can be accounted for and removed when performing data analysis (Fig. 9.2).
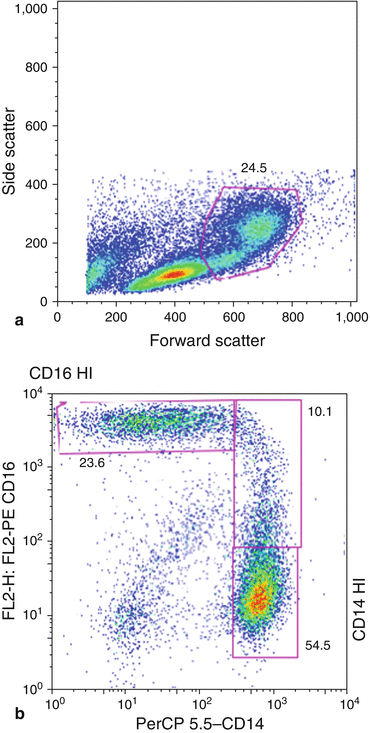
Fig. 9.2
Forward scatter vs. side scatter dot plot. Example of gating illustrated. Further analysis for evaluation of monocyte subpopulation (CD14+, CD16+)
Future Innovations
The majority of conjugated antibodies are with excitable fluorochromes at the 488 or 632 nm. However, recently there has been an increase in commercially available fluorochromes across a larger portion of the spectrum. Ideally, this should have little overlap with the commercially available fluorochromes already in use [16]. For example, at one end of the spectrum—violet (450 nm)—Alexa 350 (Molecular Probes, Eugene, Oregon) and AMCA-X fluorochromes could potentially be used. Dyes in this range include AMCyan 403, Alexa 405 (Molecular Probes, Eugene, Oregon), Pacific Blue (Molecular Probes, Eugene, Oregon) and Pacific Orange (Molecular Probes, Eugene, Oregon), to name a few. Moving along the spectrum to the blue region (488 nm), fluorochromes that are available include FITC and peridinin chlorophyll protein (PerCP). Green (532 nm) and yellow-green (565 nm) portion of the visual spectrum includes dyes for PE. Finally in the red color of the spectrum (633 nm), excitable fluorochromes include allophycocyanin (APC).
Although the expansion of commercially available excitable fluorochromes across the visual spectrum is impressive in its own right, it is not for purely esoteric reasons that multiple color conjugates were developed. This is best demonstrated by the complexity of the immune system and our attempts to define the different cell subtypes in the population. As more analysis was performed on lymphocytes using standard two-color cytometer (i.e., two conjugated monoclonal antibodies), it was discovered that there was a larger heterogeneity in the lymphocyte population and that two cell-surface markers were not enough to definitely identify a lymphocyte population [27]. It is sometimes necessary to identify as many as six different markers on the T lymphocytes in order to correctly label the population. Thus, improvements in the hardware associated with flow cytometry had to evolve in order to permit the multicolor labels needed to analyze a cell population. The human immunodeficiency virus (HIV) epidemic of the 1980s provided the initial impetus to expand flow cytometry’s capabilities in its ability to identify T lymphocyte subpopulations as research demonstrated that naïve CD8+ cells were lost in HIV infection [28, 29]. De Rosa et al. demonstrated that two-color flow cytometry was not enough to demonstrate naïve T cells and that the use of three to five markers improved the ability to identify naïve T cells more accurately [27]. The advancements made in multicolor or polychromatic flow cytometry (PFC) are not exclusive to T cell populations. Advancements have been made in the humoral branch of the immune system as well; especially in the functionality of B cells and in the different stages that the B cell exists from progenitor cell to mature cell. So not only is the morphology of distinct cells able to be elucidated, but distinctions based on function can now be performed with PFC.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


