Type 3 secretion systems
Liam J. Worrall, Julien R.C. Bergeron and Natalie C.J. Strynadka, University of British Columbia, Vancouver, BC, Canada
Introduction
In 1994, the group of Hans Wolf-Watz observed that the pathogenic Gram-negative bacterium Yersinia pestis could transfer toxin proteins (effectors) directly into the cytoplasm of infected mammalian cells. Since this process was independent from other secretion pathways known at the time, it was labeled type 3 secretion (T3S) (Salmond and Reeves, 1993; Rosqvist et al., 1994). A set of genes, organized in a complex operon structure, was shown to encode for the proteins necessary and sufficient for the translocation of effectors across both the bacterial membranes, as well as the mammalian cell membrane (Michiels et al., 1991). This set of proteins was therefore called the type 3 secretion system (T3SS).
Subsequently, homologous genes were identified in many human Gram-negative pathogens such as Salmonella enterica spp., Burkholderia spp., Chlamidia spp., Pseudomonas aeruginosa, Vibrio parahaemolyticus, Shigella spp., enterohemorrhagic E. coli (EHEC) and enteropathogenic E. coli (EPEC), as well as in the plant pathogens Pseudomonas syringae, Ralstonia solanacearum and Erwinia chrysanthemi. In most instances, the occurrence of T3S was confirmed, and has been shown to be an important factor for pathogenicity (for review, see Troisfontaines and Cornelis, 2005; Coburn et al., 2007; Tampakaki et al., 2010). The delivery of effector proteins into a host cell via T3S allows the bacterium to manipulate the bacterial–host cell interaction in its favor with effectors targeting a wide variety of cellular processes including the cytoskeleton, phagocytosis, apoptosis, and the inflammatory response (see Chapter 15).
The molecular basis for T3S was discovered in 1998, when Kubori et al. (1998) observed the presence of syringe-shaped structures on the surface of Salmonella typhimurium. These possessed a morphology similar to the hook-basal body organization of the bacterial flagellum, and were shown to be composed of proteins encoded by the Pathogenicity Island 1 (SPI-1), a major pathogenicity component of Salmonella typhimurium. This assembly, referred to as the T3SS injectisome, was shown to be necessary for the translocation of effectors. Subsequently, the direct visualization of T3SS-encoded injectisomes have also been reported for Yersinia pestis (Journet et al., 2003), Shigella flexneri (Tamano et al., 2000) and EPEC (Sekiya et al., 2001). The similarity between the injectisome and the flagellum is not only morphological: confirming a phylogenetic link between the two systems, many flagellar proteins, specifically those localized at the inner membrane, are homologous to T3SS injectisome components, suggesting a common evolutionary origin (Ginocchio et al., 1994).
In this chapter, we will describe the various T3SSs present in E. coli and related Shigella strains (Chaudhuri and Henderson, 2012) (see also Chapter 7), and their role in pathogenicity. We will then review the current knowledge of the organization of the T3SS injectisome with special attention to the structural detail as well as current insights into the possible molecular mechanisms employed by these systems for efficient secretion of effector proteins (see also Chapter 15 for a review of T3SS effector function). Finally, an overview of the complex regulation implemented for T3SS assembly and secretion will be summarized. Our review will focus on the T3SS injectisome, but various proteins of orthologous function in the flagellar T3SS will also be discussed.
Type 3 secretion systems in E. coli
LEE-encoded T3SS
The locus of enterocyte effacement (LEE) is a pathogenicity island originally identified in the EPEC strain O127:H6 (McDaniel et al., 1995), and later found in most EPEC, EHEC, and atypical enteropathogenic E. coli (aEPEC) strains. It is responsible for the distinctive formation of attaching and effacing (A/E) lesions by LEE-encoding strains, requiring first adhesion of the bacteria to the host intestinal epithelium followed by rapid recruitment of various host cytoskeletal elements that ultimately create the characteristic pedestal (lesion) beneath the adhered bacterium (Donnenberg and Kaper, 1992).
In most strains, the LEE is found inserted within tRNA-encoding sites (typically selC, pheU, or pheV) (Wieler et al., 1997; Muller et al., 2009). Sequence analysis shows a lower GC content within the LEE (38.4%) compared to the average for the overall E. coli genome (50.8%) as well as homology to transposons (Donnenberg et al., 1997) that suggests a potential mode of horizontal transfer between E. coli strains (Reid et al., 2000; Sandner et al., 2001).
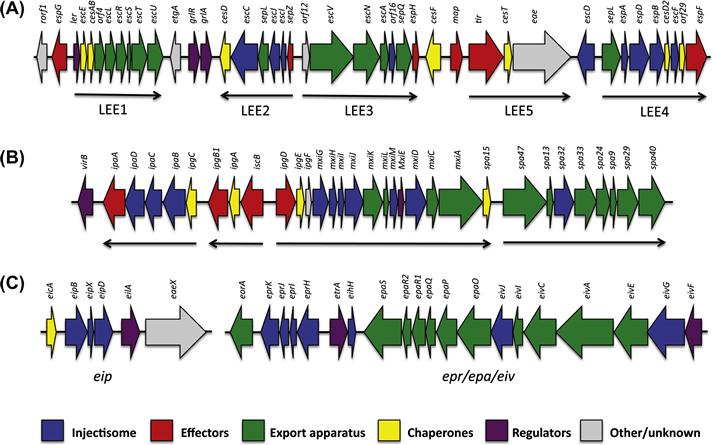
The LEE is ∼35 kb long, and contains 41 reading frames organized into five distinct operons (Figure 14.1A) (Elliott et al., 1998), which include genes encoding for several T3SS effectors and their chaperones, a T3SS injectisome and export apparatus, T3SS regulators, and an adhesin (Jarvis et al., 1995). Secretion of various T3SS effectors in EPEC-infected epithelial cells has been observed (Kenny et al., 1996; Lai et al., 1997), directly confirming that the T3SS is functional. Additionally, deletion of individual LEE genes results in a lack of A/E lesion formation and reduced virulence (Rosenshine et al., 1992; Kenny and Finlay, 1995; Tacket et al., 2000).
Evolutionary, the T3SS encoded in the EPEC LEE is most closely related to the Salmonella typhimurium SPI-2 T3SS, as determined by sequence similarity (Hensel et al., 1998), which is active only in the Salmonella-containing vacuole. It remains to be seen if this phylogenetic proximity corresponds to similar features between these two systems, or if it simply reflects a common origin.
The Mxi/Spa system of Shigella
The main determinant of virulence in pathogenic Shigella flexneri is the presence of the large virulence plasmid pWR100 (Sansonetti et al., 1982, 1983), a 200 kb plasmid encoding for approximately 100 genes (Venkatesan et al., 2001). It is found in all pathogenic serotypes of S. flexneri, S. dysenteriae and S. sonnei (Yang et al., 2005), and is necessary for pathogenicity. Importantly, this plasmid can confer invasive properties to non-pathogenic E. coli strains (Sansonetti et al., 1983) and can be transmitted between bacteria (Sansonetti et al., 1982).
Within this plasmid, a 31 kb pathogenicity island was identified (Sasakawa et al., 1988), and sequence analysis showed that it contains 34 genes organized in two clusters transcribed in opposite directions (Figure 14.1B). Subsequent studies demonstrated that it forms a functional T3SS (Menard et al., 1994), labeled the Mxi/Spa system. It is one of the best-conserved regions of the virulence plasmid (Yang et al., 2005) and is necessary for epithelial cell invasion and macrophage killing (Sansonetti, 1991; Zychlinsky et al., 1992).
The Mxi/Spa system is closely related to the well-studied inv system, the Salmonella typhimurium SPI-1 encoded T3SS (Troisfontaines and Cornelis, 2005), which is responsible for the invasion into epithelial cells, suggesting a similar role for these two T3SSs.
The ETT2 system
More recently, sequence for a putative second T3SS was identified in the genome of the EHEC strain O157:H7 Sakai (Ohnishi et al., 2000), the source of a food-borne E. coli outbreak in Japan in 1999. This system, called ETT2 (E. coli Type Three secretion 2), was subsequently found in many EPEC and EHEC strains (Hartleib et al., 2003; Makino et al., 2003). Deletion of the ETT2 reduces pathogenicity (Ideses et al., 2005; Yao et al., 2009), but the molecular aspects of this effect remain unclear. In particular, secretion has not been observed to date for this system.
The ETT2 is formed by two separate operons, located in distinct loci of the genome (Ren et al., 2004): the epr/epi/eiv operon contains 19 genes, coding for putative elements of the injectisome, while the eip locus contains six genes including a putative translocon, effector and regulator (Figure 14.1C), related in sequence to the inv/mxi/spa systems including the Salmonella typhimurium SPI-1 and Shigella T3SSs. However, sequence comparison reveals that in most strains, many mutations and sequence deletions are present, leading to the proposition that this system is non-functional (Ren et al., 2004). Nonetheless, expression of several ETT2 genes, under the control of the putative regulator, has been reported (Sheikh et al., 2006).
Structure and organization of the T3SS injectisome
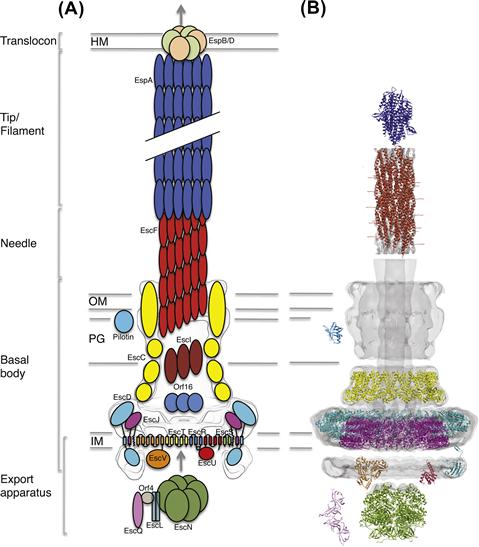
One of the most striking aspects of T3S is the encoded secretion system, a ∼3.5 MDa macromolecular apparatus often referred to as the ‘injectisome’. Since its first observation in Salmonella typhimurium (Kubori et al., 1998), our understanding of the T3SS structure and assembly has expanded through a combination of structural and genetic studies. It is composed of >20 different proteins, many of them in a highly oligomerized state. Electron microscopic (EM) analysis from various species shows a syringe-shaped structure (Figure 14.2) with a central hollow channel approximately 30 Å wide (Marlovits et al., 2004; Schraidt and Marlovits, 2011), constituting the proposed path through which T3S effectors are translocated.
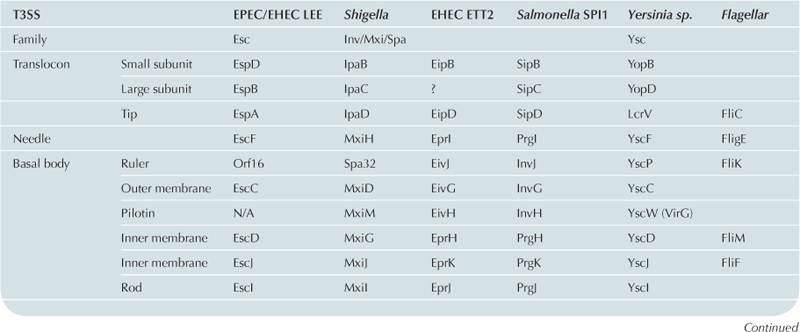
For historical reasons, the nomenclature used to name the genes of the T3SS has not been standardized. As a consequence, the names of individual proteins vary between different T3SSs (see Table 14.1 for T3SS nomenclature from different systems). In order to simplify this chapter, the EPEC LEE T3SS nomenclature will be used for general description of individual proteins.
The basal body
The basal body of the injectisome refers to the large substructure spanning from the bacterial cytosol through to the inner and outer membranes of the bacterium (see Figure 14.2). The precise definition of what constitutes the basal body varies somewhat in the published literature; here, we will use this term to describe the structural components spanning both bacterial membranes and the periplasmic space, excluding the needle projecting outward as well as the inner-membrane export apparatus. High-resolution EM reconstruction of the basal body from the Salmonella SPI-1 T3SS (Marlovits et al., 2004, 2006; Schraidt and Marlovits, 2011), as well as that from Shigella (Blocker et al., 2001; Hodgkinson et al., 2009) have revealed they are composed of several concentric, symmetrical rings, resembling the basal body found in the flagellum.
The inner-membrane region of the basal body in the EPEC T3SS is composed of two proteins, EscD and EscJ, forming an intimately associated pair of 24-mer oligomeric rings (Yip et al., 2005b; Schraidt et al., 2010; Schraidt and Marlovits, 2011). By analogy with PrgK in the Salmonella SPI-1 system (Marlovits et al., 2004; Sanowar et al., 2009; Spreter et al., 2009; Schraidt et al., 2010), EscJ is presumed to form the innermost ring component and, given the lipoprotein signal peptide at the N-terminus (Crepin et al., 2005), is localized to the inner membrane by a lipidation anchor (orthologs of EscJ in several T3SSs also possess an additional transmembrane helix at the C-terminus) (Kimbrough and Miller, 2000; Schuch and Maurelli, 2001). The EscJ structure consists of two independent small α/β domains of similar fold, joined by a structured linker (Crepin et al., 2005; Yip et al., 2005b). Based on studies of the Salmonella SPI-1 ortholog of EscD, PrgH, this protein likely forms a 24-mer ring in intimate contact with the EscJ ring in the periplasm (Sanowar et al., 2009; Schraidt et al., 2010; Schraidt and Marlovits, 2011). PrgH contains a cytoplasmic domain and a periplasmic domain, which are separated by a transmembrane helix. The cytoplasmic domain possesses a Forkhead-domain associated (FHA) fold (McDowell et al., 2011; Barison et al., 2012; Lountos et al., 2012), and has been proposed to interact with elements of the export apparatus (Johnson et al., 2008). Whether this interaction is phosphorylation-dependent, as predicted from the FHA fold, remains unknown. The periplasmic region of PrgH is composed of three domains, each possessing a similar fold to the two domains of EscJ (Spreter et al., 2009). Collectively, the dual inner membrane ring system of the T3SS is proposed to act as a structural foundation for recruitment/stabilization and final assembly of the cytosolic ATPase and several multispan membrane proteins that create an energized export apparatus for chronological secretion of downstream T3SS components and effectors (see below for further details on the export apparatus components).
The outer-membrane ring is composed of a single protein, EscC, which belongs to the secretin family of outer-membrane channels (see Korotkov et al., 2011 for review). Its stoichiometry remains controversial, with 12-mers reported for the Shigella T3SS secretin MxiD (Hodgkinson et al., 2009), 13-mers for the Yersinia T3SS secretin YscC (Burghout et al., 2004b), and 15-mers for the Salmonella SPI-1 T3SS InvG (Schraidt and Marlovits, 2011). This discrepancy may reflect true system-to-system variation or may correspond to inherent plasticity in the oligomerization state of this component, particularly when isolated in vitro. High-resolution structural information on the transmembrane domain is lacking, but the structure of the periplasmic domain from the EPEC secretin EscC has been reported (Spreter et al., 2009); it is composed of two domains N0 and N1, also found in secretins from unrelated systems (Korotkov et al., 2009). The N1 domain possesses a similar fold to the domains of the periplasmic regions of EscJ and PrgH. Intriguingly, domains possessing this fold were also found in other oligomeric assemblies (Korotkov et al., 2009; Levdikov et al., 2012; Meisner et al., 2012), leading to the suggestion that this fold is a common ‘ring-building motif’ (Spreter et al., 2009).
In most T3SSs, a lipoprotein termed the pilotin promotes outer-membrane targeting, insertion and oligomerization of the secretin component (Crago and Koronakis, 1998; Burghout et al., 2004a). Pilotins are presumed to act through a concerted binding of the C-terminal end of the secretin and interaction with lipids in the membrane (Okon et al., 2008; Ross and Plano, 2011); however, the pilotins of different systems do not show any measurable sequence conservation, and even their overall structure is not conserved (Lario et al., 2005; Izore et al., 2011; Koo et al., 2012). Of note, a pilotin has not yet been identified in the EPEC T3SS, and the sequence of the corresponding secretin, EscC, appears to lack a C-terminal pilotin-binding domain, suggesting that this protein utilizes an alternative mechanism for its assembly in the outer membrane (Gauthier et al., 2003).
In addition to these structural components, analysis in the Salmonella SPI-1 system suggests the conserved but less well-characterized proteins PrgJ (EscI in EPEC) and InvJ (EscP in EPEC) are also essential components of the generalized T3SS basal body, with both proteins co-purifying with isolated injectisomes in vitro (Marlovits et al., 2004, 2006). These proteins are localized to the periplasmic region of the basal body, and are proposed to interact with the secretin and needle T3SS components, as well as with components of the inner membrane export apparatus (Kimbrough and Miller, 2000; Sukhan et al., 2003; Sal-Man et al., 2012). EscI (PrgJ in the SPI-I T3SS) is hypothesized (although not yet verified) to be a polymerized hollow rod that spans the T3SS basal body to form a continuous path with the downstream needle component (Marlovits et al., 2006; Sal-Man et al., 2012). This so-called ‘inner-rod’ has also been implicated in the transition from secretion of the needle protein to tip and translocon proteins (EspABD in EPEC), a process known as substrate switching (discussed below) (Marlovits et al., 2004; Sal-Man et al., 2012). NMR studies of a refolded, recombinant form of the EscI ortholog PrgJ shows that this protein largely lacks a defined structure (Zhong et al., 2012), although the relevance of this observation within the assembled injectisome remains to be determined. The biochemical role of InvJ/EscP is better characterized: its deletion creates T3SS variants with abnormal needle length (Tamano et al., 2002; Journet et al., 2003; Mota et al., 2005; Marlovits et al., 2006; Monjaras Feria et al., 2012), leading to the hypothesis that this protein acts as a ‘molecular ruler’ to regulate T3SS needle formation, although the mechanism by which such measurement is achieved remains unexplained.
The extracellular components: the needle, filament, and translocon
The T3SS needle is the portion of the apparatus extending away from the extracellular surface of the basal body; it is a ∼70 Å wide superhelical structure, with a ∼30 Å hollow channel in the middle (Figure 14.2), and varies in length depending on the system (∼23 nm for the EPEC T3SS (Monjaras Feria et al., 2012), ∼45 nm for the Shigella T3SS (Tamano et al., 2002)). The needle is composed of multiple copies of a single protein (EscF in EPEC), which is well conserved between species. EM helical reconstructions of the needle from the Shigella (MxiH) (Cordes et al., 2003; Fujii et al., 2012) and Salmonella SPI-1 (PrgI) T3SSs (Galkin et al., 2010) suggested that the needle helical arrangement possesses a pitch of 25 Å, and contains approximately 5.5 subunits per turn, which is similar to the flagellum filament.
Purified needle proteins spontaneously form extended needle-like oligomeric assemblies of several µm in length (Poyraz et al., 2010), which has impaired high-resolution X-ray crystallographic characterization in the context of the assembled needle; nonetheless, several structures of point and truncation mutants that inhibit oligomerization, or of needle monomers complexed with their specific chaperone (T3S encoded proteins that prevent premature polymerization of newly expressed needle components within the bacterial cytosol), have been reported (Deane et al., 2006; Zhang et al., 2006; Quinaud et al., 2007; Sun et al., 2008; Poyraz et al., 2010; for review, see Blocker et al., 2008). In all cases, a similar two-helix coiled-coil bundle, linked by a conserved turn defined by a PxxP sequence motif is observed. Recently, elegant studies of the Salmonella typhimurium needle using solid-state NMR (Poyraz et al., 2010; Loquet et al., 2012) has allowed for an accurate model of its assembled structure, providing atomic-resolution insights into the orientation of individual PrgI molecules within the needle, and their interaction with one another (Loquet et al., 2012).
Importantly, the needle not only forms a hollow conduit to passage effectors from the bacteria to the infected host, but has also been implicated in additional roles including mammalian cell sensing, and, along with the inner rod, in substrate switching during various stages of injectisome assembly and virulence effector secretion (Kenjale et al., 2005; Davis and Mecsas, 2007).
At the distal, extracellular end of the needle, multiple copies of a terminating tip protein have been reported in various species (EspA in EPEC, IpaD in Shigella); EM studies suggest the tip is pentameric, matching the cross-sectional symmetry of the needle (Deane et al., 2006; Broz et al., 2007; Epler et al., 2012). The proposed role of the tip is to sense the presence of mammalian cells, through direct interaction with membrane lipids (Olive et al., 2007; Veenendaal et al., 2007), as well as to serve as an adapter between the needle and the downstream translocon pore that inserts directly into the host cell membrane (Picking et al., 2005). The structures of the tip protein from several systems (Derewenda et al., 2004; Erskine et al., 2006; Johnson et al., 2007) show the presence of a core domain forming a two-helix coiled-coil similar to the needle protein. In addition, a small N-terminal helical domain regulates binding to the needle, requiring partial unfolding for the final assembly of the needle/tip complex (Wilharm et al., 2007; Lunelli et al., 2011). A third domain, found at the C-terminus, has been proposed to be involved in contacts with components of the translocon (Roehrich et al., 2010).
Of note, in EPEC and related species, an unusual ortholog of the T3SS tip protein, EspA, has been shown to be solely responsible for formation of the filament, a highly extended polymeric structure that protrudes from the bacteria, with lengths in excess of 600 nm (Knutton et al., 1998; Daniell et al., 2001; Sekiya et al., 2001). These extended translocation filaments in EPEC and EHEC are thought to help span the significant mucosal layer of infected epithelial cells in the host gut. EM helical reconstructions of sheared EspA filaments (Wang et al., 2006) shows a structure similar to the T3SS needle, with approximately 5.5 proteins per turn. Variations in helical parameters observed in these high-resolution EM maps suggest a potential conformational lability or ‘breathing’, proposed to help accommodate the significant cellular forces likely experienced at the bacterial/host interface and potentially to help propel virulence proteins internalized within these hollow filaments along their considerable lengths to the translocation pore at the host cell membrane. Recombinant, purified EspA spontaneously oligomerizes into filament-like structures, and the crystal structure of EspA in complex with its T3SS encoded anti-polymerization chaperone CesA (Yip et al., 2005a) revealed the canonical two-helix coiled-coil domain, suggesting an oligomerization mechanism similar to that of the needle, as well as an unstructured region between these two domains that is likely surface-exposed.
The T3SS translocon is a pore-forming complex proposed to insert directly into the eukaryotic host cell membrane, allowing T3S effector proteins entry into the cytoplasm (for review see Mattei et al., 2011). The pore is a proposed hetero-dimeric complex composed of two integral membrane proteins (EspB, EspD in EPEC), which interact with the tip structure at the apical end of the needle (Dorman, 2004). Like the needle and tip components, the translocon proteins are exported by the injectisome itself, a process likely initiated upon host-cell contact (Enninga et al., 2005). EspD is predicted to contain a two transmembrane region and a coiled-coil domain (Dasanayake et al., 2011) while EspB has one transmembrane spanning segment, a coiled-coil domain, and an additional amphipathic helix (Luo and Donnenberg, 2011). Presumably the coiled-coil domains in both pore components facilitate interactions with the preceding needle tip/filament proteins.
At present there is limited structural information for the translocon, hindered by poor expression of this pore-forming complex using standard recombinant methods. Low-resolution EM and AFM analysis of the EPEC translocon in transfected sheep red blood cell membranes appears to suggest a non-symmetrical six to eight subunit hetero-oligomeric stoichiometry (Ide et al., 2001) although it remains to be confirmed if the structures observed in this study are indeed the translocon. The translocon proteins have so far been recalcitrant to higher-resolution structural study, however Barta et al. recently published the X-ray crystallographic structures of the protease-resistant domain of the small subunit from Shigella (IpaB) and Salmonella SPI-1 (SipB) components (Barta et al., 2012). The structures revealed a coiled-coil motif with observed conservation between the various species (despite the relatively low sequence identity of ∼ 22%). Further, structural comparisons revealed similarity to other bacterial pore-forming proteins, notably colicin Ia, suggesting an evolutionary relationship.
Prior to secretion, the two hydrophobic translocon proteins are stabilized within the bacterial cytoplasm by a common T3SS chaperone, CesD (classified as a type II chaperone; for a recent review of T3SS chaperone architecture/function see Wilharm et al., 2007). The localized interaction interface of the translocon proteins with their cognate chaperone has recently been characterized, revealing independent binding to a common surface on the chaperone. Although these studies are beginning to illuminate our understanding of translocon function, many critical questions such as oligomerization state and stoichiometric make-up of the pore and the consequent effect on translocon insertion into the host membrane and effector secretion remain unanswered.
The inner-membrane export apparatus
The T3SS inner-membrane export apparatus consists of five integral membrane proteins (EscRSTUV), predicted to localize within the dual inner-membrane rings (EscJ/EscD) at the base of the basal body, along with several cytoplasmic membrane-associated proteins including the T3SS ATPase (EscN; see Figure 14.2). It should be noted that these proteins, which regulate secretion across the inner membrane, have been termed the export apparatus in the literature to distinguish them from the distinct and downstream translocating pore (the translocon) that passages substrate through the host membrane. The export apparatus proteins are amongst the most highly conserved proteins of the T3SS and have closely related homologs in the flagellar T3SS. EscRSTU are encoded on the same operon whilst the larger EscV is encoded separately (Figure 14.1A). Given their membrane-spanning nature, it is perhaps not surprising that little is currently understood at the molecular level regarding the coordinated function of the export apparatus, although it is generally believed to be central to recruitment and regulation of initial insertion of effectors into the injectisome (for review of the related flagellar export apparatus see Minamino et al., 2008b). Presumably, packing of EscRSTUV within the EscJ/D rings maintains sufficient room for passage of downstream needle/translocon and effector proteins through the inner membrane, as suggested by recent high-resolution EM studies in Salmonella typhimurium (Schraidt and Marlovits, 2011). The significant cytoplasmic domains of two of the export apparatus proteins, EscU and EscV, have been suggested to form a functional export gate controlling access to the inner channel (Saijo-Hamano et al., 2010). The soluble ATPase is thought to dock to this export gate (Minamino et al., 2012), where it uses the energy derived from ATP hydrolysis to dissociate effectors from their cognate chaperones prior to insertion into the inner-membrane channel.
Structural models for soluble domains of the export apparatus proteins have begun to emerge in recent years. EscV is the largest of the export apparatus proteins (∼70 kDa) with eight predicted TM helices and a large C-terminal cytoplasmic domain (∼40 kDa). X-ray crystallographic analysis of the analogous domain has recently been published for the Salmonella SPI-1 T3SS ortholog InvA (Lilic et al., 2010; Worrall et al., 2010) in addition to three structures of the flagellar homolog FlhA (Bange et al., 2010; Moore and Jia, 2010; Saijo-Hamano et al., 2010). These structures show a highly conserved overall fold consisting of four subdomains. Of interest, the structures capture the protein in different conformational states with variation in the degree of opening of a central cleft, although the functional relevance of this is at present unclear. The cytoplasmic domain of FlhA has been shown to bind subunits destined for the formation of the flagellar filament suggesting a role in substrate recognition and regulation of assembly (Bange et al., 2010; Diepold et al., 2011). Using a fluorescent tag, Cornelis and colleagues demonstrated that the Yersinia homolog YscV co-localizes with other T3SS proteins in multiple copies, unlike the other export apparatus proteins, suggesting it forms higher-order oligomers (Diepold et al., 2011).
EscU is predicted to contain two TM helices and a C-terminal ∼20 kDa cytoplasmic domain. EscU orthologs have also been implicated in the temporal regulation of injectisome assembly, acting as part of a molecular ‘switch’ that regulates the chronological secretion of apparatus components and virulence effectors. It has been demonstrated that flagellar and non-flagellar homologs undergo a spontaneous autocleavage event in their C-terminal cytoplasmic domain leaving a cleaved fragment still associated with the C-terminal domain (Minamino and Macnab, 2000a; Lavander et al., 2002). The recent structures of the cytoplasmic domain of EscU along with orthologs from Salmonella typhimurium (SpaS), Yersinia enterocolitica (YscU) and Shigella (Spa40) show a small mixed alpha/beta domain (Deane et al., 2008a; Zarivach et al., 2008; Lountos et al., 2009; Wiesand et al., 2009). The structures revealed the molecular basis for an intein-like autocleavage reaction via asparagine cyclization on a highly conserved surface exposed loop, resulting in the remodeling of surface features and concomitant changes in electrostatics in the EscU cytosolic domain. Non-cleavable mutants show aberrant secretion of the tip or translocator proteins suggesting cleavage is required for their recognition and a role in switching from translocon to effector secretion (Zarivach et al., 2008).
In addition to the membrane-embedded components of the export apparatus, several membrane-associated proteins are also involved in the regulation of secretion, notably the T3SS ATPase EscN. The identification of an ATPase associated with the flagellar T3SS (Vogler et al., 1991) and subsequently a homolog in the T3SS injectisome of Yersinia (Woestyn et al., 1994) led to the proposal that it was the energy source for protein translocation. More recently however, several studies have suggested that the proton-motive force (PMF), the electrochemical potential difference of protons across a membrane, provides the energy for protein unfolding and translocation (Minamino and Namba, 2008; Paul et al., 2008) supported by the observation that secretion can still occur, albeit less efficiently, in absence of the ATPase in both the flagellar T3SS (Minamino and Namba, 2008) and the T3SS injectisome (Wilharm et al., 2004). Based on the ability of the Salmonella SPI-1 T3SS ATPase to dissociate chaperone–effector complexes in an ATP-dependent manner (Akeda and Galan, 2005), it is currently believed that the ATPase is required for the effective targeting of T3S chaperone–effector complexes to the base of injectisome and subsequent effector release and initial unfolding prior to translocation. One intriguing feature of the ATPase is the similarity to the F/V/A-ATPases, rotary motors that couple transmembrane ion flow to ATP catalysis, first documented some 20 years ago (Vogler et al., 1991; Woestyn et al., 1994). Confirming the close structural similarity, structures of the EPEC T3SS ATPase EscN (lacking the N-terminal domain) (Zarivach et al., 2007) and the flagellar ATPase FliI (Imada et al., 2007) were published several years ago. FliI crystallized as a homo-dimer conserved in nature to the hetero-dimer found in the F/V/A-ATPases and experimental evidence has confirmed that, like the F/V/A-ATPases, the type 3 ATPases likely function as hexamers in the physiological context (Kazetani et al., 2009). Initially reported for the flagellar T3SS, this evolutionary relationship has been extended to further soluble export apparatus proteins: firstly, the flagellar T3SS FliH protein family, which was found to bind and regulate the activity of the ATPase (Minamino and Macnab, 2000b; Blaylock et al., 2006), and has significant sequence similarity to the peripheral stalk of the F/V/A-ATPases (Pallen et al., 2006), which acts as a stator to prevent rotation of the catalytic subunits. Secondly, structural similarity has also been observed for the small flagellar T3SS coiled-coil protein FliJ and the central stalk of the F/V/A-ATPases (Ibuki et al., 2011), which transduces the rotational force between the membrane-embedded and catalytic components. Both FliH and FliJ form complexes with the flagellar T3SS ATPase and have orthologs in the T3SS injectisome (EscL family and EscA family, respectively). The detected evolutionary relationship between the soluble export apparatus components of the T3SS and the F/V/A-ATPases, and the involvement of the PMF in T3SS transmembrane translocation, a pivotal feature of F/V/A-ATPases, supports a possible common evolutionary origin and related mechanism of these two transmembrane molecular machines.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


