Comparative genomics of pathogenic Escherichia coli
Jason W. Sahl1, Carolyn R. Morris2 and David A. Rasko2, 1Translational Genomics Research Institute, Flagstaff, AZ, USA, 2University of Maryland School of Medicine, Baltimore, MD, USA
Introduction
Escherichia coli is a human gut commensal isolate and deadly human pathogen (Kaper et al., 2004). E. coli is easily cultured from the human gut and has been the focus of scientific studies for greater than one hundred years (as will be discussed elsewhere in this book). The availability of clinical, laboratory, and commensal isolates, as well as the associated clinical/epidemiological data have provided highly characterized isolates for whole genome sequencing.
The first Escherichia coli genome sequenced was the laboratory-adapted isolate, K12 MG1655 (Blattner et al., 1997). The single chromosome consisted of approximately 4.6 Mb in sequence that encodes approximately 4300 genes. At the time of sequencing, 38% of all coding regions had no predicted function. The sequencing of this isolate was rapidly followed by the publication of genomes from O157:H7 isolates EDL933 (Perna et al., 2001) and Sakai (Hayashi et al., 2001). Comparisons were made between these genomes with the genome of K12 to determine the genetic variability between isolates in the same species. In 2002, the genome of the uropathogenic isolate, CFT073, was completed (Welch et al., 2002). Comparisons among the three sequenced isolates at that time demonstrated that all isolates only shared ∼39% of all coding regions. At the time, the low conservation of genes and coding regions in a single species changed the existing paradigm of gene conservation. Early thoughts on genome sequencing were that ‘representative isolates’ could be sequenced and they would represent the species or in this case pathovar. This concept was rapidly discarded in light of the low level of conservation in this species.
In 2008, the first confirmed intestinal commensal isolate (HS) was published (Rasko et al., 2008). A pan-genome analysis, based on peptide identity and conservation, was conducted on 17 E. coli isolates, including eight new genomes, sequenced at that time. The results demonstrated that the conserved genomic core of E. coli consists of ∼2200 genes. The analysis of representatives of multiple pathovars demonstrated that few pathovar-specific genes were identified. It was hypothesized that the low level of conservation was in part due to the strain selection bias that attempted to include as broad a collection of isolates as possible. These early studies highlighted the diversity in the species and pathovars.
In 2009, Touchon et al. published a paper that performed a pan-genome analysis on a larger number of sequenced genomes (n = 20) (Touchon et al., 2009). The analysis demonstrated that the number of core genes in E. coli remained around 2000. The analysis demonstrated the size variability in the E. coli chromosome, from 4.6–5.3 Mb, and the variability in plasmid content between isolates. Variation in the pan-genome conserved core between these two studies could be attributed to using different bioinformatic methodologies, as well as defining the term ‘core pan-genome’ differently. While these variations in the absolute numbers are small, both studies highlight the fact that approximately only half of the E. coli genome is highly conserved in all isolates examined. At the time these studies were published this was an interesting finding that impacted the definition of species.
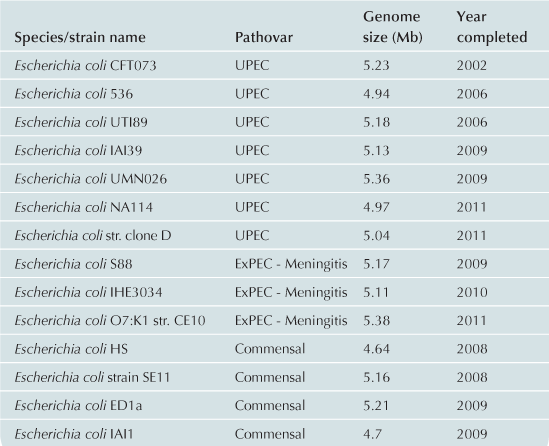
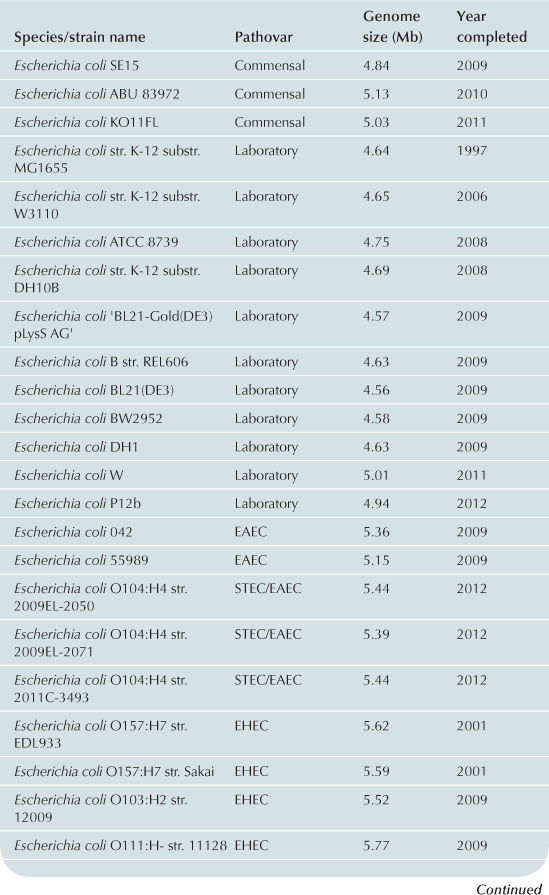
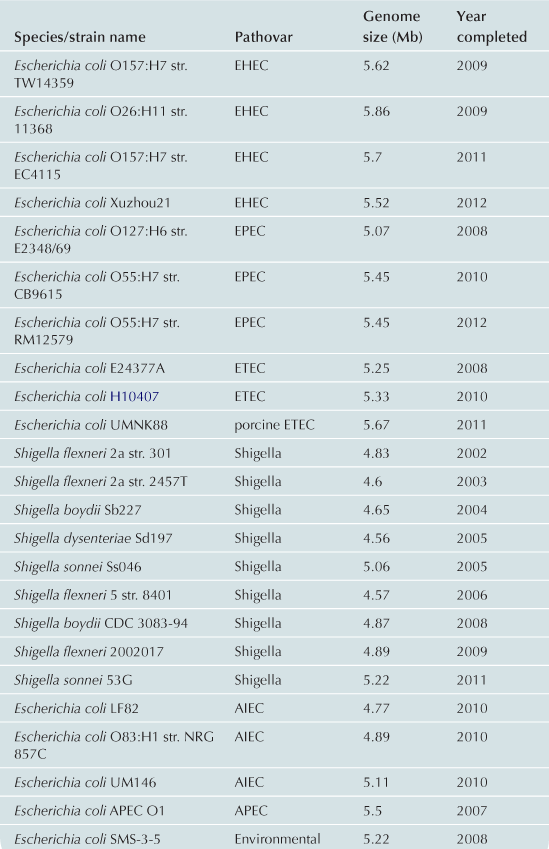
To date approximately 60 E. coli/Shigella genomes have been completed (Table 2.1) with representatives from each of the pathovars. Recent advances in bench-top next generation sequencing platforms have provided independent investigators with the ability to sequence isolates of E. coli. As of September 2012, there are >1000 registered E. coli sequencing projects available in Genbank (http://www.ncbi.nlm.nih.gov/). These sequencing projects may further clarify the evolutionary history of the species, the diversity of gene content within the species, and identify genes associated with pathogenesis within each pathovar. Ideally, the ongoing genomic and phylogenomic studies will identify regions of the genome that are not encoded on mobile elements, and thus may represent more stable and effective biomarkers for each pathovar. Additionally, the sequencing will provide deeper insight into genomic diversity, combined with studies of the transcriptome, will provide clues into the regulatory networks of these pathogens.
This chapter will be separated into descriptions of the genomic studies that have been completed on isolates on each of the specific pathovars.
Uropathogenic E. coli
Uropathogenic E. coli (UPEC) are thought to be innocuous in the gastrointestinal tract, but become pathogenic in the urinary tract (Chen et al., 2006; Schwartz et al., 2011). High-throughput sequencing has shed light on the diverse genomic organization and distribution of virulence factors in this pathotype. The UPEC are the prototypes of the extraintestinal pathogenic E. coli (ExPEC) isolates and often people refer to UPEC isolates as ExPEC isolates, but in this case we will only discuss the UPEC, as the majority of the sequencing has focused specifically on the UPEC.
The first UPEC isolate, CFT073, was sequenced, as a collaborative effort, by Welch et al., in 2002. CFT073 was isolated from the blood of a patient with pyelonephritis; the genome consists of 5.2 Mbp, 5533 protein-coding genes, and no identified virulence plasmids. This strain is considered to be the prototype UPEC isolate, but the isolation from the blood of a patient suggests that it may be particularly pathogenic in comparison to other UPEC isolates. UPEC have acquired virulence genes that allow them to survive, and possibly thrive, in the GI tract but result in a disease presentation in the urinary tract. These genes are often found on horizontally acquired pathogenicity islands (Hacker et al., 1992; Hacker and Kaper, 2000). The CFT073 sequence confirmed the presence of previously described pathogenicity associated islands (PAIs) located at tRNA genes pheV, pheU, and asnT. PAI-pheV contains genes for pili associated with pyelonephritis (pap), aerobactin synthesis, hemolysin (hly), capsule synthesis, and two autotransporters. PAI-pheU also contains pap genes as well as a siderphore receptor (Welch et al., 2002). PAI-asnT is similar to the high pathogenicity island of Yersinia pestis and contains yersiniabactin genes (Perry and Fetherston, 2011). The location and composition of the PAIs in CFT073 were surprisingly different from the known PAIs in two other well-studied UPEC isolates 536 and J96 (Swenson et al., 1996; Middendorf et al., 2001). The PAI-pheV of J96 also contains hly but additionally encodes heat resistance hemmagglutanin (hra) and cytotoxic necrotizing factor (cnf-1). J96 and 536 both encode yersiniabactin at PAI-asnT, but 536 has three additional PAIs at selC, leuX, and thrW. In 536, pap and hly are located at PAI-selC, a second copy of hly and P-related fimbriae genes (prf) are found at PAI-lueX, and S-type fimbrial adhesion (sfa) and iron siderphore (iro) genes are located at PAI-thrW. In addition to the PAI encoded genes, CFT073 also has several potential virulence factors located outside of PAIs (Welch et al., 2002). These include seven putative auto-transporters and 12 putative fimbriae (10 chaperone-usher and two type IV).
In 2006, two additional UPEC isolates were sequences. UTI89 was sequenced by Chen et al. (2006) at Washington University. UTI189 was isolated from the urine of a patient with cystitis and the genome consists of 5.1 Mbps and one plasmid, pUTI89, which is 114 230 bps. There are 5066 predicted protein-coding genes and four large PAIs. In contrast, Brzuszkiewicz et al. (2006) sequenced UPEC strain 536, isolated from the urine of a patient with pyelonephritis. The 536 genome contains 4.9 Mbps, 4747 predicted coding sequences, and no plasmids. Comparison of five E. coli genomes revealed 432 genes that were present in UPEC strains 536 and CFT073 but not in EDL933, Sakai, or laboratory strain K12 MG1655; this analysis suggested that these genes may have a role in urovirulence. Additionally, 427 genes were identified that were present in 536 but absent in all other published E. coli genomes (Brzuszkiewicz et al., 2006). Many of these genes are found in PAIs. PAI-selC encodes hyl and two sets of fimbriae genes; PAI-leuX contains another copy of hyl and prf; PAI-thrW contains sfa and iro genes; PAI-asnT contains yersiniabactin genes; and PAI-pheV contains capsule genes. By creating PAI deletion mutants they found PAI-selC, –leuX, and –asnT all contributed to virulence in a mouse model of ascending UTI, while only deletion of PAI-selC and PAI-leuX together had an impact on virulence in a mouse model of urosepsis. This result suggested that there are many factors that contribute to UTI establishment, but hly, which is found in two copies on PAI-selC and PAI-leuX is associated with the later stages of urosepsis.
In 2007, Lloyd et al. described ten new genomic islands in CFT073, in addition to the three previously described PAIs, by comparative genome hybridization of 10 E. coli strains (Lloyd et al., 2007). In 2009, Lloyd et al. compared nine genomic island mutants in a mouse model of ascending UTI. PAI-metV and PAI-aspV mutants were out-competed by wild-type CFT073 in the bladder and genes c3405-c3409 of PAI-metV were found to be important for colonization of both bladder and kidneys and specific to UPEC. PAI-aspV was further dissected, and contact-dependent inhibition gene cdiA and the autotransporter protease gene picU were determined to be important for colonization of the bladder. The RTX family exoprotein A gene, tosA, was important for colonization of the kidneys. PAI-aspV additionally contains a copy of the ferric binding protein (fbp) operon, which is also present in genomic island cobU. Deletion of both copies of the fbp operon was required for attenuation. This work emphasized that no single factor was responsible for virulence, even within a single strain, and that multiple genes and genomic regions contribute to the fitness of CFT073 in the urinary tract.
Later in 2009, two more UPEC strains, UMN026 and IAI39, were sequenced by Touchon et al. (2009). UMN026 was isolated from the urine of a patient with cystitis. The genome contains 5.2 Mbps with 4918 protein-coding genes, and two plasmids; one plasmid was 122 kb with 149 protein-coding genes and the other was 34 kb with 54 protein-coding genes. IAI39 was isolated from the urine of a patient with pyelonephritis. The genome consists of 5.1 Mbps, 4906 protein-coding genes, and no plasmids. This group compared UMN026, IAI39, and the three previously published UPEC genomes with 15 other diarrheagenic or commensal E. coli strains (Touchon et al., 2009). When considering intrinsic extraintestinal virulence (in a mouse model of bacteremia) (Johnson et al., 2006), the authors were not able to identify any single genes that were specific to a virulent phenotype (Touchon et al., 2009). This demonstrated that the distinct pathogenic potential of UPEC, as compared to other E. coli, was not the result of a fixed group of virulence factors, but rather a variable collection of factors.
More recently, in 2010, Hagan et al. published the first transcriptional profile of pathogenic E. coli taken directly from patients with a naturally occurring infection (Hagan et al., 2010). Isolates were collected from the urine of eight women with bacteriuria, and the transcriptomes were compared by microarray to the same isolates grown statically in sterilized urine. When these data were compared to gene expression data from a mouse model (Snyder et al., 2004), there was an overall positive correlation. Iron acquisition genes and metabolic genes were most strongly correlated with colonization, toxin genes were moderately correlated, but fimbriae and adhesion genes were poorly correlated. Multiple fimbriae genes are highly expressed by CFT073 in mice, including the fim operon. Despite being highly expressed in mice, only two of six isolates tested from humans expressed the fim operon in the urine, even though seven of the eight strains were confirmed to make functional fimbria. These findings demonstrate that while the mouse model largely reflects the transcriptional profile of UPEC in humans, there are some differences, and continued study of UPEC transcriptomics may lead to more effective therapeutics (Hagan et al., 2010).
New approaches to studying global changes in gene expression, such as RNAseq, will further our understanding of UPEC pathogenesis. While genomic analyses have demonstrated that there is variable collection of virulence factors that confer pathogenic potential to UPEC isolates, the coordinated expression of these factors is also likely to influence pathogenesis. Further study of UPEC transcriptomics may identify novel virulence factors and targets for vaccine and therapeutic development.
Shiga-toxin producing E. coli/enterohemorrhagic E. coli (STEC/EHEC)
The Shiga toxin, encoded by a lambdoid prophage in E. coli isolates (Campbell et al., 1992), was first characterized in an epidemic case of dysentery caused by Shigella dysenteriae (Trofa et al., 1999). Shiga toxin-producing E. coli (STEC) have also been described and are associated with a range of symptoms in the human host, from mild diarrhea to severe hemorrhagic colitis (see Chapter 5) (Kaper et al., 2004). Most importantly, STEC are associated with hemolytic uremic syndrome (HUS) (Corrigan et al., 2001), which differentially affects young children in developing countries. The E. coli outbreak in Europe in the summer of 2011 was caused by an isolate that had the Shiga-toxin phage in the context of a phylogenetic group that did not previously harbor this genetic element (Frank et al., 2011; Rasko et al., 2011). If strict molecular definitions are observed, the outbreak isolate could be classified as an STEC, but not EHEC, as the majority of the genome was most similar to an enteroaggregative E. coli (see below). These findings from genomic studies highlight the limitations with regard to inferred phylogeny of the current typing schema that rely on mobile genomic regions as biomarkers or small amounts of DNA for typing.
The STEC isolates can be segregated into groups that either contain the Locus of Enterocyte Effacement (LEE) (McDaniel et al., 1995) pathogenicity island (Kaper et al., 2004) and those that do not. The LEE encodes a type III secretion system that injects infectors into the host cell that results in the formation of attaching and effacing lesions (see Chapters 5, 6, 14, and 15) (Donnenberg and Kaper, 1992; McDaniel et al., 1995). LEE-positive STEC are frequently termed enterohemorrhagic E. coli (EHEC), due to the frequent manifestation of hemorrhagic colitis in infected hosts. LEE-positive isolates can also be classified as attaching and effacing E. coli (AEEC), which also includes enteropathogenic E. coli (EPEC, see below). Thus there is some ambiguity in how these isolates that can be phylogenetically related (Figure 2.1) are classified based on the presence or absence of specific molecular markers. As with the other pathovars, it is hoped that continued sequencing will identify stable regions of the genome that can be utilized as potential biomarkers for rapid and accurate classification of these pathogens. It is possible that in some cases, as with the EHEC/STEC division, that multiple markers will be required.
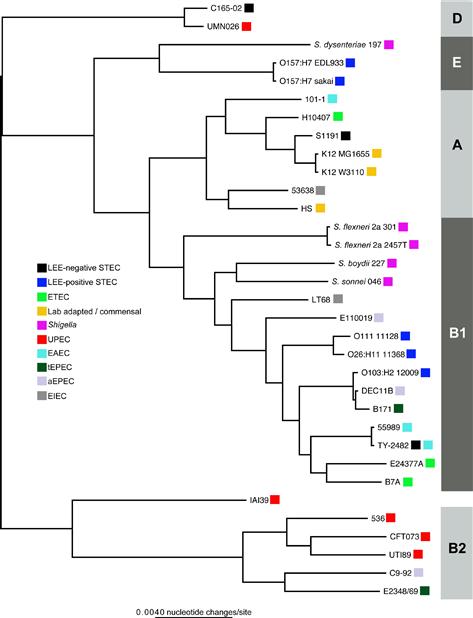
FIGURE 2.1 A phylogenetic tree inferred from 2.5 Mb of genomic sequence conserved in all isolates. The alignment was parsed from a Mugsy (Angiuoli and Salzberg, 2011) whole genome alignment of all genomes. A phylogeny was inferred on this concatenated alignment with FastTree2. Boxes to the right of each genome indicate the corresponding pathotype.
The first EHEC genome sequenced was O157:H7 strain EDL933, which was isolated from an outbreak in Michigan (Perna et al., 2001). A comparison of this genome with K12 identified ∼1400 new genes associated with virulence factors, prophages, and genes associated with variable novel metabolic pathways. This publication was followed closely by a manuscript that described the sequence of O157:H7 strain Sakai (Hayashi et al., 2001), which was isolated from an outbreak in Japan (Watanabe et al., 1996). The results of this comparison identified 1632 proteins present in the Sakai strain and absent in K12. These two isolates from the same pathovar provided the data for the first intrapathotype comparisons (Kudva et al., 2002). These studies provided evidence of greater intrapathotype diversity than was previously recognized and signaled that the evolutionary processes leading to the creation of these pathogens were not likely to be simple, linear or easily understood (see Chapter 3).
Outbreaks of O157:H7 infection, such as the spinach-associated outbreak in 2006 have occurred in developed countries, including the United States (Parker et al., 2012). An analysis of 25 O157:H7 genomes associated with three foodborne outbreak events identified 1225 single nucleotide polymorphisms (SNPs) in this clonal expansion (Eppinger et al., 2011). In fact, within the O157:H7 clade, variation in virulence (Manning et al., 2008) has been associated with specific clades in the clonal complex. Whole genome sequence data have been used to not only characterize the evolution of virulence of this important pathogen, but will help in the surveillance of this pathogen.
In addition to O157:H7 EHEC isolates, genomes have also been sequenced from other STEC/EHEC serotypes; these genomes include those of O111:H-strain 11128, O26:H11 strain 11368, and O103:H2 strain 12009 (Ogura et al., 2009). A phylogeny of 345 orthologous coding regions demonstrated that the non-O157 EHEC isolates are not contained on a single phylogenetic branch (Ogura et al., 2009). However, a phylogenetic tree based on the entire gene repertoire demonstrated that all EHEC genomes could be grouped together, most likely due to the presence of common secreted effectors and LEE-associated genes (Ogura et al., 2009).
Other LEE-negative STEC isolates have also been associated with severe diarrheal symptoms and HUS (Johnson et al., 2006; Newton et al., 2009; Rasko et al., 2011). Recently nine LEE-negative STEC isolates were sequenced and comparative analysis demonstrates the extreme phylogenetic diversity of this group (Steyert et al., 2012). This study of LEE-negative STEC has revealed that these isolates are even less similar than the LEE-positive STEC, suggesting a diverse evolutionary history. One would expect this finding considering the typing schema is based on a mobile element that can insert into most E. coli genomes. Additional detailed comparative analysis demonstrated the diversity of secreted effectors and stx insertion sites in the group. The major finding in the study was that the Shiga-toxin phage and the genomic backbone are not intimately linked, but some features of the phage define the location of insertion (Steyert et al., 2012).
The genomic examination of STEC and EHEC isolates highlights the juxtaposition of the current typing schema; while the virulence factors are important for the clinical treatment and identification of pathogens, these features do not always concur with the phylogenetic relationships of the isolates. The advent of the new sequencing technologies will allow rapid identification of whole genome sequencing into the clinical paradigm and then it will be possible to characterize isolates by their genomic content, and not phenotypic presentation; problems of associating phenotype with gene content may be avoided.
Enteropathogenic E. coli (EPEC)
Enteropathogenic E. coli (EPEC) are traditionally classified by the presence of the LEE and the absence of stx genes encoding Shiga toxins (Kaper, 1996). As with EHEC, there are subclassifications within EPEC based on virulence factor presence and absence. Isolates that contain the LEE region and the EPEC adherence factor (EAF) plasmid (Kaper et al., 2004) encoding genes for a bundle forming pilus (BFP) (Giron et al., 1991) are frequently termed typical EPEC (tEPEC), while LEE-positive, BFP-negative isolates are classified as atypical EPEC (aEPEC) (Kaper et al., 2004). The bundle forming pilus creates a network of filaments that bind the bacteria together into what is known as a microcolony. Both tEPEC and aEPEC have been associated with diarrheal disease (Trabulsi et al., 2002), which suggests that the BFP is not absolutely required for EPEC virulence. Indeed, volunteer studies demonstrate that loss of the EAF plasmid or mutation of bfp genes reduces, but does not eliminate pathogenicity (Levine et al., 1985; Bieber et al., 1998). Phylogenetic analysis based on concatenated multilocus sequence typing (MLST) genes demonstrated that tEPEC are found in two primary lineages (EPEC1 and EPEC2), although several smaller clades have been identified (Orskov et al., 1990; Lacher et al., 2007). This diversity suggests that the acquisition of LEE and the EAF plasmid have occurred on multiple, independent occasions.
The genomic landscape of EPEC is as follows: The first two EPEC genomes sequenced were B171 and E110019 (Rasko et al., 2008); B171 is a tEPEC isolate from the EPEC2 lineage (Giron et al., 1991) and E110019 is an aEPEC that was the etiological agent of a large outbreak in Finland in 1990 (Viljanen et al., 1990). A comparative analysis revealed that although 200 unique genes were identified in the two EPEC genomes, compared to other sequenced genomes, few (n = 9) pathovar-specific genes were identified (Rasko et al., 2008). The prototype EPEC isolate E2348/69, which is part of the EPEC1 clade, was sequenced in 2009 (Iguchi et al., 2009). A comparative analysis demonstrated greater than 400 unique genes in this isolate. Furthermore, only 21 secreted effectors were identified in the genome of E2348/69 compared to approximately 50 described in the genomes of O157:H7 isolates (Tobe et al., 1999).
The diversity of EPEC has been previously determined by the typing of intimin variants (Lacher et al., 2006); the intimin gene (eae) is encoded by the LEE. In a study of 151 eae-positive isolates, 26 distinct profiles were observed using fluorescent restriction fragment length polymorphism. Phylogenetic analyses of EPEC have typically been performed from concatenated MLST markers (Lacher et al., 2006). Phylogenies inferred from whole genome sequence data have demonstrated that trees inferred from concatenated MLST markers do a poor job at recapitulating the whole genome phylogeny (Leopold et al., 2011; Sahl et al., 2011). Thus whole genome phylogeny will help better understand the phylogenetic history of EPEC isolates in the broader context of all E. coli and Shigella genomes.
The sequencing of additional EPEC genomes, as part of the GSCID project (http://gscid.igs.umaryland.edu/wp.php?wp=emerging_diarrheal_pathogens), has provided additional information on the evolution of EPEC. For example, some BFP-positive EPEC are closely related to BFP-negative EPEC phylogenetically (Hazen et al., 2012); this may be due to the loss of the EAF plasmid prior to or during lab passage. Previous studies have demonstrated that in E2348/69, the prototype EPEC isolate, the plasmid is very stable (Levine et al., 1985; Donnenberg et al., 1993). However, as more isolates are sequenced and investigated, the existing dogma is increasingly challenged. For example, based on a PCR screen analysis, BFP-positive and BFP-negative isolates would be classified as tEPEC or aEPEC, respectively, despite potentially sharing a closely related common ancestor. To incorporate phylogenetic information into a clinical assay, comparative genomics were used to identify genomic markers that distinguish between the different lineages of EPEC. A multiplex PCR reaction was designed to detect classical virulence factors as well as phylogenetic markers (Hazen et al., 2012). This methodology will define a new paradigm in which whole genome sequence data focuses better diagnostics to understand both virulence profiles and phylogenetic history.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


