Chapter 12 Tumours
Incidence and mortality
The last 100 years have seen the incidence of lung cancer transformed from it being an almost unknown disease to it far outstripping all other forms of cancer as a cause of death in many western countries. The death rate from lung cancer exceeds 60 per 100 000 in Great Britain and is close to this in many developed countries. In British men, it is responsible for about 35% of cancer deaths, and about 8% of all deaths, whilst in women it now rivals breast cancer as the major cause of cancer mortality. In many developed countries the incidence in men has levelled off and in some it has started to decline, but in women it is still rising,1 the lag being almost totally due to women taking to smoking later than men. The male-to-female ratio was nearly equal in England and Wales at the end of the nineteenth century but climbed to 6 by 1951.2 Now it is again verging on equality as deaths in men decline slightly and deaths in women rise at a rate comparable to that seen in men in the first half of the twentieth century. Furthermore, for a given level of smoking women are at higher risk of developing lung cancer than men.3–5 A higher level of aromatic DNA adducts (indicating DNA damage) is reported in the lungs of female smokers.6
Many developing countries have a relatively low incidence of lung cancer but are beginning to see the rises already experienced elsewhere as the inhabitants take to smoking in increasing numbers. There are also substantial racial differences in susceptibility to the carcinogenic effect of cigarette smoke. For example, at any level of smoking black Americans have a risk of lung cancer 1.8 times greater than that of their white compatriots.3 It is likely that in the twenty-first century there will be a significant shift in the geographical distribution of lung cancer.
Lung cancer is largely a disease of later life. In patients younger than 40 years of age there is a lower male-to-female ratio and a higher proportion of adenocarcinoma but as in the elderly cigarette smoking is the most important cause.7 The tumour is very rare in children but occasional cases are encountered.8
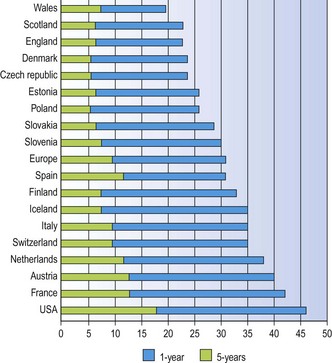
Lung cancer carries a very poor prognosis. Surgical survival figures may reach 46% (and nearly 60% for T1N0M0 tumours)9 but they are based on a highly selected group of patients. Spread of the tumour often results in it being inoperable at presentation while many patients with lung cancer also have coexistent chronic obstructive lung disease that renders them unfit for surgery. Overall 5-year survival rates vary from 6% to 14%, the variation being mainly attributable to national differences in access to specialist care (Fig. 12.1.1).10–12 Note also has to be taken of the significant number of patients who suffer recurrence more than five years after apparent eradication of their tumour; 11% in one study.12a
Aetiology
Smoking
The evidence linking cigarette smoking and lung cancer was first established in the early 1950s when the smoking habits of large groups of patients with lung cancer were compared with those of matched controls. These provided clear evidence that the majority of patients with carcinoma of the lung were heavy cigarette smokers and that the incidence of the disease in non-smokers was low.13–15 Later prospective studies, in which the incidence of lung cancer was determined over a long period of time in large groups of people with different smoking habits, were even more conclusive. One of the best known of these studies was carried out in the UK on a group of 40 000 doctors. In 1951, each completed a questionnaire asking for details of their smoking habits. By 1964 there was ample evidence of a clear link between cigarette smoking and deaths from lung cancer.16,17 Similar studies were carried out in the USA with identical conclusions.18,19 These studies established that the risk of lung cancer is increased in smokers compared with non-smokers by a factor of 10. Pipe smokers and cigar smokers have a significantly lower incidence than cigarette smokers, probably due to them not inhaling their smoke to the same degree.20 The risk increases with the number of cigarettes smoked and is proportional to the length of time a person has smoked, being greatest in those who commenced at an early age.18 The term ‘pack years’ is often used to quantify this combination of risk factors, ‘1 pack year’ equating to 20 cigarettes per day for 1 year or 40 per day for 6 months. In those who stop smoking, the risk begins to decline immediately but it takes about 15 years for the risk in ex-smokers to approach that of non-smokers.16 The incidence of lung cancer is increased in those with other smoking-related diseases, such as chronic bronchitis.21
The risk of lung cancer is reduced by switching to filter tip and low-tar cigarettes,22 and from cigarettes to cigars or pipes.23 However, it is suggested that the switch to low-tar cigarettes has led smokers to inhale more deeply and thereby contributed to the increased incidence of peripheral tumours.24 There is no clear evidence that French cigarettes, which contain air-cured tobacco, are any safer than those made of flue-cured tobacco, but the resultant smoke is more alkaline and the contained nicotine can be absorbed from the mouth. Consequently, fewer French than British cigarette smokers admit to inhaling their smoke and this may contribute to the observed lower incidence of lung cancer in France than the UK, but there are other differences between the two countries: dietary differences are mentioned below.
Passive smoking, which is exposure to the cigarette smoke exhaled by others and the smoke emitted by their smouldering cigarettes (referred to as mainstream and sidestream smoke respectively), carries a small risk of lung cancer.25,26 Compared with a non-smoker, the increased risk of lung cancer is generally quoted as a percentage for a passive smoker but so-many-fold for an active smoker, the respective figures being in the order of 25% and 13-fold (which equate to 1.25-fold versus 13-fold). A comparison of nicotine metabolite levels suggests that passive smoking entails an exposure equivalent of up to 1 cigarette a day. It is estimated that passive smoking accounts for one extra death from lung cancer or heart disease for every 10 000.
Numerous carcinogens have been identified in cigarette smoke. The more important ones are listed in Box 12.1.1. Many of them are thought to act by binding to DNA. Of particular note are the polycyclic hydrocarbons found in the neutral fraction of the particulate phase and the nitrosamines of the basic fraction. The former has been linked to squamous cell carcinoma and the latter to adenocarcinoma.27 There appears to be genetic susceptibility to the carcinogenic effects of these chemicals, which is possibly based on differences in the amounts of activating enzymes in the lung, many of which are P450 cytochromes.28
Box 12.1.1 Major carcinogens of tobacco smoke
Particulate phase
| Neutral fraction | Benzpyrene |
| Dibenzanthracene | |
| Benzofluoranthenes | |
| Basic fraction | Nitrosamines |
| Residual fraction | Nickel |
| Cadmium | |
| Polonium-210 |
Direct evidence of the effects of cigarette smoke on bronchial epithelium is provided by pathological studies, which demonstrate a close association between metaplasia and atypia and the number of cigarettes smoked.29,30 Long-term animal experiments have been difficult to institute but have provided further supportive evidence of the link between tobacco smoke and lung cancer.31,32
Cigarette smoking predisposes to pulmonary carcinoma of all the major histological types, but the link is strongest with squamous cell and small cell carcinoma, which generally arise in central airways, and weakest with adenocarcinoma, which is commoner in the periphery of the lung.33,34
Environmental carcinogens
As with constituents of cigarette smoke, many inhaled chemicals are not hazardous as such but are transformed to reactive intermediates within the lungs by enzymes such as N-acetyl transferase, glutathione-S-transferase and members of the P450 family. After allowing for differences in smoking habits it is found that the risk of lung cancer is higher in urban areas than in rural areas, suggesting a role for general atmospheric pollution.35,36 Major atmospheric pollutants include polycyclic hydrocarbons from the combustion of fossil fuels. Domestic coal fires were formerly the prime source of polycyclic hydrocarbons in the UK and it was estimated that in the 1950s air pollution accounted for about 10% of lung cancer in men.37 Since that time coal fires have been eliminated in most major conurbations and concentrations of benzpyrene have fallen dramatically. The main source in the UK, as in many countries, is now motor vehicle exhaust and particular attention is being paid to diesel smoke. Some epidemiological studies have identified an increased risk of lung cancer in occupational groups having heavy exposure to diesel exhaust, notably one that examined American maintenance workers exposed to fumes from diesel locomotives.38 A review by an international agency concluded that there was evidence that diesel engine exhaust was carcinogenic in experimental animals and that there was limited support for this in humans.39 There appears to be only a weak link between urban nitrogen oxide and sulphur dioxide pollution and lung cancer.40
Ionising radiation
Similar increases in lung cancer incidence were seen in North American miners exposed to radon and its daughter products41,42 and such exposure probably accounts for the increased incidence seen in English haematite miners.43 The increase involves both squamous cell carcinoma and small cell carcinoma, but particularly the latter.44,45 Cigarette smoking is an important cofactor . The increased risk is 67 times greater in heavy smokers exposed to heavy doses of irradiation than in non-smokers with comparable radiation exposure, indicating a multiplicative effect, as seen with asbestos.46,47
Radon levels may also reach dangerous levels within the home; the degree of danger depends on the character of the underlying rock.48,49 It is difficult to prevent radon seeping into buildings and easier to instal solid floors over a radon sump that can be exhausted to the exterior. Many countries have policies to control indoor radon, usually focusing on buildings where radon levels exceed a certain level, although there is evidence that in some countries it would be cost-effective if basic preventive measures were required for all new homes.50
There is also an increased incidence of lung carcinoma in patients given therapeutic irradiation, for example to the spine for ankylosing spondylitis51 and to the breast for mammary carcinoma.52,53 Survivors of the Hiroshima and Nagasaki nuclear bombings were also at increased risk of lung cancer.54,55
Asbestos
An association between exposure to asbestos and carcinoma of the lung was first demonstrated conclusively in 1955.56 As with mesothelioma, the association is stronger with amphibole asbestos than chrysotile, but with all forms of asbestos much higher levels of exposure are required to produce carcinoma than mesothelioma. All histological types are involved but the proportion of adenocarcinoma is increased.57–59 Cigarette smoking is an important cofactor. In a study of 17 800 asbestos workers the risk in smokers was 14 times that in non-smokers.60 Table 7.1.7 (p. 349) provides further data on this, showing that the lung cancer risk for workers heavily exposed to asbestos is 5 times that of unexposed persons whatever their smoking status, while the risk for smokers is 11 times that of non-smokers whether they are exposed to asbestos or not. As a consequence, the lung cancer incidence among smokers exposed to asbestos is 53 times higher than that among unexposed non-smokers – a multiplicative rather than additive effect.61 The risk diminishes in those who cease smoking. A practical preventive point arising from this is that most of the excess risk can be abolished by eliminating or decreasing exposure to either asbestos or cigarette smoke. Also, it suggests that asbestos acts as a promoting agent rather than a primary carcinogen. The vexed question of whether asbestos causes lung cancer in the absence of asbestosis is considered on page 349. Asbestos fibre burdens are generally low in lungs resected because of cancer, indicating that asbestos is not a major cause of lung cancer in the general population.62,63
Other industrial chemicals
Apart from asbestos and ionising radiation, six other agents encountered in industry are accepted pulmonary carcinogens: arsenic, chloromethyl ethers, chromium, nickel, polyaromatic hydrocarbons and vinyl chloride. Many others, including cadmium, formaldehyde and dioxin, are suspected of having a similar effect (Box 12.1.2).64 Cigarette smoking is often an additional factor.65
Box 12.1.2 Occupational agents associated with lung cancer, categorised by the criteria of the International Agency for Research on Cancer64
Workers in the coal gas industry exposed to polyaromatic hydrocarbons have a lung cancer risk twice that of the general population,66 and lung cancer has also been associated with a range of metal-refining and smelting processes: nickel, chromium, arsenic and cadmium have all been incriminated.67,68 In the 1930s a higher incidence was reported amongst workers at the Clydach nickel refinery in south Wales. Changes in the process later resulted in a reduction of the risk but the hazard remains in several other nickel refineries.69 Exposure to both nickel and cadmium was related to lung cancer in workers in a battery plant.70 Poorly soluble hexavalent chromium pigments cause lung cancer but very soluble chromium compounds are comparatively safe.71,72
Arsenic and its compounds are associated with skin and lung carcinomas, an increased incidence being found in people administered arsenic therapeutically, in vineyard workers involved in spraying arsenical insecticides, and in copper smelters exposed to arsenic.73 Arsenic may also be responsible for an increased risk of lung cancer identified in antimony workers.74 It is also anticipated that the next decade will witness widespread arsenic-induced cancer in Bengal and Bangladesh following the provision of water free of bacterial contamination by extraction from shallow aquifers. Up to 10 million shallow tube wells have been dug and it is estimated that 1 million of these are heavily contaminated with naturally occurring arsenic released by anaereobic metal-reducing bacteria.75 At least 100 000 cases of arsenic-induced skin growths have been identified and it seems inevitable that cancer of the skin and respiratory tract will follow.
Exposure to the alkylating agent chloromethyl methyl ether, which is used as a cross-linking agent in ion exchange resins and as an intermediate in the production of organic chemicals, carries an increased risk of lung cancer, particularly if there is contamination with bis(chloromethyl) ether.76 Small-cell carcinoma is particularly increased.77
Epidemiological studies have also incriminated silica exposure as a risk factor for lung cancer, and as with asbestos there is uncertainty as to whether the risk is due to high silica levels alone or to the fibrotic process.78 Claims for compensation are therefore more likely to succeed if silicosis as well as carcinoma can be demonstrated.
Diet
Many of the differences in disease incidence seen between Mediterranean and northern European countries have been ascribed to diet, especially the differing intake of antioxidants. Dietary differences are probably important in regard to cardiovascular disease but may also affect cancer incidence. Many cigarette smokers have therefore gleefully increased their consumption of red wine on evidence that is to date at best equivocal.79
Vitamin A deficiency leads to squamous metaplasia of mucosal surfaces, dryness of the skin, eyes and mouth, and to an increased susceptibility to cancer.80 Cigarette smoke augments the metaplastic action of vitamin A deficiency in rats,81 whilst in humans low vitamin A intake leads to an increased incidence of lung cancer in smokers.82 Folate is another dietary component that protects against squamous metaplasia.83 Diets high in carotenoid-rich fruits and vegetables are associated with a reduced risk of lung cancer, as are high serum levels of vitamin E and beta-carotene, but long-term dietary supplementation with these substances either had no effect or resulted in a slightly higher incidence of lung cancer in male smokers.84–86
Pulmonary fibrosis
Idiopathic pulmonary fibrosis is associated with a 14-fold increased incidence of lung cancer,87–89 similar to that noted above in asbestosis.60 In both these diseases, the tumours are of all histological types and they arise more frequently in the periphery of the lower lobes where the fibrosis is most marked.90,91 Although the original publications suggested that adenocarcinoma was the most common histological type, it is now believed that the cell-type distribution is similar to that in the general population.58 Sarcoidosis has been reported to convey an increased risk of lung cancer in some studies92,93 but not in others.94
In contrast to these forms of diffuse pulmonary fibrosis, the role of localised scars in the development of lung cancer is now questioned. Many adenocarcinomas show pronounced pleural puckering due to central scarring (see Fig. 12.1.23, p. 554) and have often been assumed to represent ‘scar cancers’, meaning that the scar preceded and predisposed to the cancer. However, it is often impossible to tell whether the tumour developed in a pre-existing scar, and can therefore be designated a true scar cancer, or whether the central scar is secondary to the tumour.95 Nevertheless, there is considerable evidence, both radiological and pathological, supporting the view that the scar is the product of the tumour rather than the reverse. Relevant radiological observations96 include the following:
Pathological observations97–101 supporting the view that central scars are usually a product of the tumour rather than the reverse include the following:
Irrespective of whether the scar or the tumour comes first, central scarring in a pulmonary adenocaricoma is an adverse prognostic indicator, being associated with increased vascular and pleural invasion and lymph node metastasis.97,102
Viruses
Viral oncogenes participate in carcinogenesis by the RNA of the viral genome being transcribed into the DNA genome of the host by the enzyme reverse transcriptase. The DNA sequences transcribed by viral oncogenes are virtually identical to sequences in the cellular DNA of most animal species and presumably have the same potential for affecting malignant growth as cellular oncogenes. Viruses have been suggested in the past as being related to the development of some lung cancers, and the application of molecular probes has given some impetus to such investigations. Papillomavirus of types 6 and 11 is commonly found in tracheobronchial papillomatosis and in the rare papillary squamous carcinomas that complicate papillomatosis103 while type 18 has been identified in a small proportion of the more common non-papillary squamous cell carcinomas of the bronchus.104 Particularly notable is an exceptionally high rate of human papillomavirus 16- and 18-positive lung cancers in non-smoking Taiwanese women.105 Epstein–Barr virus has been shown to be associated with bronchopulmonary as well as nasopharyngeal lymphoepitheliomas but with only occasional lung tumours of the more common histological types.106,107 There is an increased incidence of lung cancer in acquired immunodeficiency syndrome (AIDS) but this is not thought to be directly attributable to the human immunodeficiency virus.108
Heredity
The great majority of smokers reaching old age do not die of lung cancer, suggesting that individuals differ greatly in the way they metabolise xenobiotic substances.109 These differences are presumably inborn. Genetic differences could influence the risk of cancer in several ways.110 They could affect the enzymatic elimination or activation of carcinogens,111,112 or the expression of oncogenes or tumour suppressor genes (see below). Epidemiological studies have shown an excess of lung cancer in close relatives, identifying an approximately twofold-increased risk of lung cancer in those with a family history of the disease.113–115 Various genes conferring increased susceptibility to lung cancer have now been identified.116,117 Those located on chromosomes 6 and 15 are particularly notable, chromosome 15 being the site of the nicotinic acetylcholine receptor gene cluster. The susceptibility these genes confer appears to be unrelated to smoking status or propensity to smoke tobacco.118 The role of genetic factors in the development of lung cancer is dealt with more extensively below, under pathogenesis.
Malformations
The cystic type 1 congenital pulmonary airway malformation is often accompanied by focal mucous cell hyperplasia that occasionally undergoes malignant transformation, usually to a mucinous adenocarcinoma of predominantly lepidic pattern (see p. 59), but the common hamartoma (see p. 626) is not a precursor of bronchopulmonary carcinoma.119–121
Pathogenesis
Premalignant changes evident morphologically
Squamous metaplasia and dysplasia
The regenerative capability of bronchial epithelial cells has been extensively studied. Hyperplasia of the surviving basal cells is an early regenerative event that is followed by the development of a stratified squamous epithelium.122 Further differentiation into mucous cells and thence ciliated cells follows but mucous differentiation is dependent upon the availability of vitamin A.81,123 The application of carcinogens such as benz-pyrene, asbestos or nitrosamines results in the proliferation of bronchial neuroendocrine cells, which may undergo squamous metaplasia.124–126 It is clear therefore that basal, mucous, neuroendocrine and metaplastic squamous cells are closely related and readily transform one to another. This marked metaplastic potential of the bronchial epithelium provides no support for facile suppositions regarding the histogenesis of particular types of lung carcinoma from particular precursor cells.
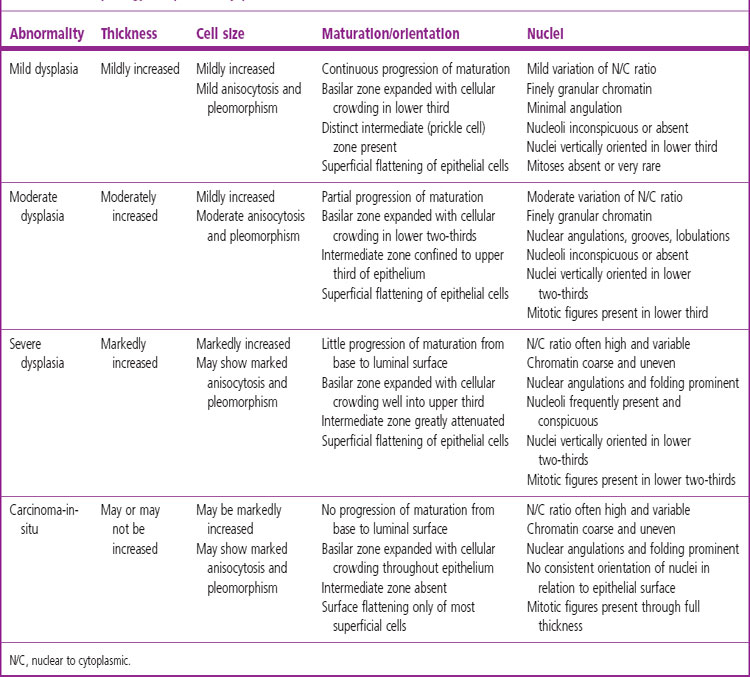
The sequence of hyperplasia–metaplasia–dysplasia–carcinoma-in-situ–invasive carcinoma (Fig. 12.1.2) is well documented in human airways127–129 and a grading system, which details the histological features evident in the surface epithelium, has been introduced (Table 12.1.1).130 Changes are also found in the mucosal blood vessels. Several groups have described hyperplastic capillary loops in close apposition to dysplastic epithelium, giving rise to the term ‘angiogenic squamous dysplasia’.131 The vascular changes can be detected by fluorescence bronchoscopy, suggesting that it may be possible to eradicate the lesions before they become invasive.132
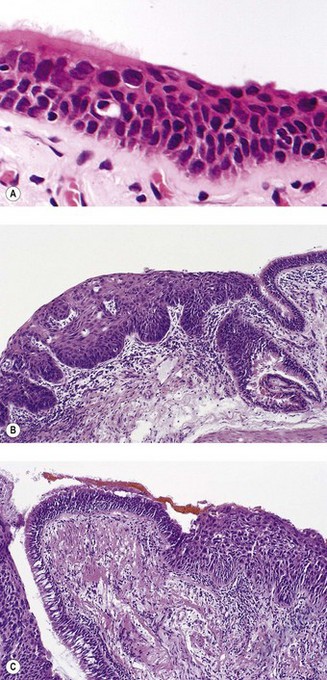
Uranium miners in the USA provided cytopathologists with the opportunity of studying the gradual evolution of bronchial carcinoma. Periodic sputum examination showed squamous metaplasia followed by gradually increasing cellular atypia in those miners who later developed invasive carcinoma of either squamous or small cell type. Atypical cells could be identified in the sputum for many years prior to the development of invasive tumours: on average, dysplastic changes lasted 8 years and carcinoma-in-situ a further 4 years before there was invasion (Table 12.1.2).127 Isolated foci of in situ squamous cell carcinoma amenable to limited surgical procedures are occasionally identified133 but at presentation invasion has generally supervened.
Table 12.1.2 Evolution of invasive squamous cell carcinoma in American uranium miners followed by periodic sputum examination127
| Average age at onset (range) | Average duration (years) | |
|---|---|---|
| Mild atypia | 48 years (38–59) | 2.89 |
| Moderate atypia | 51 years (34–67) | 3.23 |
| Marked atypia | 55 years (38–70) | 2.23 |
| Carcinoma-in-situ | 57 years (37–70) | 4.04 |
| Invasive carcinoma | 61 years |
Further evidence of dysplasia preceding carcinoma came from a large autopsy study in which the whole bronchial tree was examined in a series of patients dying of lung cancer.29 This showed widespread premalignant changes involving mucosa that appeared macroscopically normal, including foci of carcinoma-in-situ and occasional microinvasive cancers distant from the main tumour.
Foci of hyperplasia, metaplasia and dysplasia are also frequently seen adjacent to invasive tumours in surgical specimens,134 and similar changes have been described in experimental dogs exposed to cigarette smoke.31 Full-thickness squamous metaplasia is not necessary before atypical features are seen in the proliferating basal cells; dysplasia may be present beneath an intact surface layer of columnar cells.29 The transition from normal bronchial epithelium to squamous epithelium is usually abrupt. Atypical epithelium may extend deeply into bronchial glands replacing duct and acinar lining cells, resulting in appearances that mimic microinvasive carcinoma, but the basement membrane as yet remains intact.
The risk of squamous metaplasia and dysplasia progressing to invasion has been assessed by following patients with these lesions by periodic bronchoscopy. One study extending for up to 7 years put the risk at 4% for squamous metaplasia, 9% for low-grade dysplasia and 32% for high-grade dysplasia.135 Another extending over a similar period found that low-grade dysplasia did not progress at all but that the risk of progression in severe dysplasia/carcinoma-in-situ was 17%.136
In view of the extent of these premalignant changes, it is not surprising that there is a high incidence of double or second primary lung cancers.137,137a Even triple primary tumours are described.138 Using standard criteria of a double primary growth, namely involvement of different lobes, or different histological types, or a time interval over 3 years,139 it has been shown that 4% of lung cancer patients have more than one lung tumour at presentation and that a further 6% develop a second primary lung growth later.137,137a The patients most at risk of developing lung cancer are therefore those who have had one in the past. These patients might therefore be worth following up particularly frequently.
Patients with squamous dysplasia or carcinoma-in-situ may be asymptomatic or present with cough and haemoptysis. However, it is often difficult to know how much their cough is due to the dysplastic change rather than other smoking-related diseases such as chronic bronchitis. The epithelial changes are focal, usually developing at the bifurcations of segmental bronchi and subsequently extending proximally and distally. They are sometimes evident as a grey plaque but are not always visible to the bronchoscopist, particularly when routine white light is used. Autofluorescence bronchoscopy is more sensitive.140 Treatment is problematical because the lesions are frequently multiple. Patients are therefore often followed up by serial bronchoscopy until early invasion is apparent when the lesion may be treated by local ablation or surgical resection.141
Atypical adenomatous hyperplasia and adenocarcinoma-in-situ
The hyperplasia–dysplasia–neoplasia sequence is again encountered in the periphery of the lung, where it is important in the development of non-mucinous adenocarcinomas,142,143 particularly when there is diffuse pulmonary fibrosis.87–89 More recently it has been recognised that focal alveolar hyperplasia in the absence of fibrosis is an important precursor of pulmonary adenocarcinoma, evolving through a stage of adenocarcinoma-in-situ to invasive adenocarcinoma. The hyperplasia phase is generally known as atypical adenomatous hyperplasia,144–147 although in the past it has also been termed bronchioloalveolar cell adenoma.148,149 Adenocarcinoma-in-situ is a new term that replaces the older one of bronchioloalveolar cell carcinoma.150
Remarkably, foci of atypical adenomatous hyperplasia are quite commonly found when sought in lungs resected for cancer, yet they have evidently lain undiscovered by generations of pathologists dealing with such specimens. Admittedly, they are generally small and easily overlooked. Their identification is facilitated by immersion in Bouin’s solution.151 They represent foci of hyperplastic type II alveolar epithelial cells or bronchiolar Clara cells. Autopsy studies of cases free of lung cancer have identified focal adenomatous hyperplasia in 3–6%,152,153 while in lungs resected for cancer atypical adenomatous hyperplasia has been reported in up to 50% of cases, with the highest figures coming from Japan.148,151,154–157 The tumour that atypical adenomatous hyperplasia most often accompanies is a non-mucinous adenocarcinoma, but it is sometimes a large cell or squamous cell carcinoma.157 Multiple foci of atypical adenomatous hyperplasia may be found, particularly in association with multiple adenocarcinomas. Molecular studies show that individual lesions differ in their genetic make-up, establishing that they are independent of each other and of any accompanying carcinoma and do not represent micrometastases of the latter.158
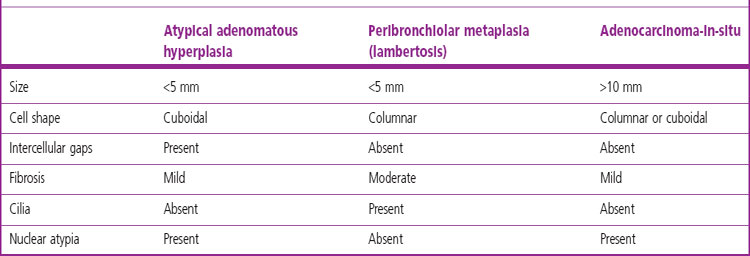
Atypical adenomatous hyperplasia is a focal lesion, generally less than 5 mm in diameter, in which the alveoli are lined by cuboidal to low columnar cells (Fig. 12.1.3), which have the ultrastructural features of type II pneumocytes and Clara cells. Ciliated and mucous cells are not a feature.130,159 The interstitium may show some fibrosis but this is generally mild. Atypical adenomatous hyperplasia is distinguished from adenocarcinoma-in-situ by its size (generally less than 5 mm whereas most carcinomas exceed 10 mm), gaps between the epithelial cells as opposed to a continuous cell line and the cells being cuboidal rather than columnar (Table 12.1.3). They have dense chromatin, prominent nucleoli and sparse cytoplasm. Mitoses are rarely evident but varying degrees of atypia are observed146,160 and the familiar hyperplasia–dysplasia–neoplasia sequence is easily envisaged. Morphometry,161,162 argyrophilic nucleolar organiser counts,163 DNA status,162,163 loss of heterozygosity164 and the identification of a range of oncogenes within the atypical lesions145,161,165–170 provide further support for this form of hyperplasia being premalignant. The demonstration of monoclonality suggests that it is neoplastic144,171 and it is now widely regarded as being the precursor of most peripheral adenocarcinomas. Nevertheless, the presence of multiple foci of atypical adenomatous hyperplasia does not appear to be related to patient survival.149,155 Patients without cancer who are found to harbour this lesion should be kept under surveillance: further intervention is not warranted.
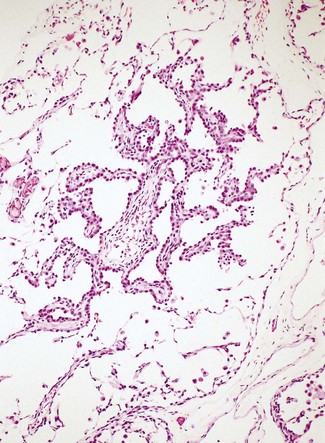
Figure 12.1.3 A focus of atypical adenomatous hyperplasia identified in a random section of lung excised because of carcinoma.
Table 12.1.3 Atypical adenomatous hyperplasia, peribronchiolar metaplasia (lambertosis, see p. 121) and adenocarcinoma-in-situ (formerly non-mucinous bronchioloalveolar cell carcinoma)
As mentioned above, adenocarcinoma-in-situ is a new term that replaces bronchioloalveolar carcinoma.156 Its new name reflects better its non-invasive nature. The lesional cells grow along the alveolar walls without destroying them (Fig. 12.1.4), a pattern of growth known as lepidic, meaning scale-like (as in the scales of a fish). Two forms of bronchioloalveolar carcinoma have traditionally been described, mucinous and non-mucinous, but it is now recognised that the mucinous variety almost always shows some invasive activity. Adenocarcinoma-in-situ is therefore almost always non-mucinous while the great majority of tumours formerly classified as mucinous bronchioloalveolar carcinoma are now termed mucinous adenocarcinoma of predominantly lepidic pattern. The latter is accordingly described later on page 554. Only non-mucinous adenocarcinoma-in-situ will be described here.
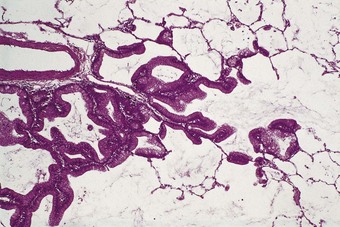
Non-mucinous adenocarcinom-in-situ predominates in smokers and is usually evident radiologically as a ground-glass opacity. It consists of Clara cells or type II pneumocytes, often in combination (Fig. 12.1.5).172,173 The Clara cells have a narrow base and a bulbous apex, resulting in a characteristic ‘hobnail’ pattern. On occasion it can be difficult to distinguish the tumour from reactive hyperplasia but a tall columnar cell shape, cellular crowding, cytological atypia and papillary infolding all favour malignancy. The presence of cilia does not exclude malignancy,174 but renders it unlikely. Immunocytochemistry and molecular studies emphasise the differences between non-mucinous adenocarcinoma-in-situ and mucinous adenocarcinoma of predominantly lepidic pattern.175 Non-mucinous adenocarcinoma-in-situ expresses the pulmonary marker thyroid transcription factor (TTF-1) but fails to stain with the colonic marker cytokeratin 20 whereas the reverse is found with mucinous adenocarcinoma of predominantly lepidic pattern.176–179 Also, non-mucinous adenocarcinoma-in-situ often displays mutation of the epidermal growth factor receptor (EGFR) gene whereas mucinous adenocarcinoma of predominantly lepidic pattern shows K-ras but not EGFR when a mutation is present.180
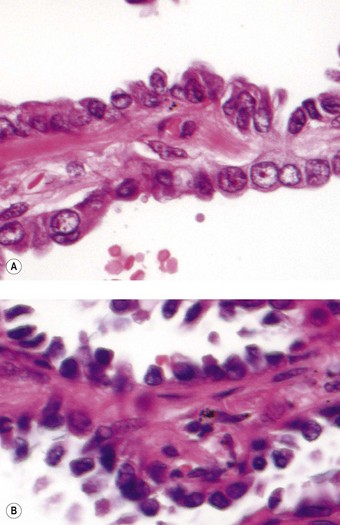
Invasive activity excludes a diagnosis of adenocarcinoma-in-situ but it can be difficult to recognise early invasion, particularly when there is alveolar collapse. The distinction is important because patients with adenocarcinoma-in-situ show 100% 5-year survival whereas invasion implies metastatic potential.181 Microinvasion, defined as invasion no greater than 5 mm, carries a prognosis intermediate between that of adenocarcinoma-in-situ and adenocarcinoma containing invasive areas greater than 5 mm across.182,183 Collapse is recognised by the lining cells being similar to those in non-collapsed areas and the spaces being regularly distributed, whereas with invasive tumour the acini vary in size and their lining cells are irregular. Solid nests or single cells also favour invasion.184 The presence of intraluminal macrophages implies continuity between the malignant cells and surrounding alveolar spaces, favouring non-invasive growth. Conversely, basement membrane stains, such as those recognising type IV collagen, facilitate the identification of invasion.182,185,186 They also demonstrate that the epithelial basement membrane is usually destroyed when there is central fibrosis.187 Not surprisingly, therefore, it has been shown that prominent central scarring as opposed to simple collapse is an adverse prognostic feature in small peripheral lung cancers.97,102,181,188 The terms Noguchi types A–C have come into use: type A is an in-situ adenocarcinoma that shows no collapse, type B an in-situ adenocarcinoma that shows collapse and type C an acinar adenocarcinoma in which invasion is detected within a central scar. It is envisaged that these represent successive stages in the evolution of a peripheral adenocarcinoma.
The multifocal micronodular hyperplasia of type II pneumocytes reported in tuberous sclerosis (see p. 701) is not recognised as being premalignant.
An infectious disease of sheep known as jaagsiekte, droning sickness or pulmonary adenomatosis closely resembles human adenocarcinoma-in-situ189 but the two are considered to be separate diseases. A report of a protein immunologically related to the jaagsiekte sheep retrovirus in some human lung carcinomas190 has not been supported by subsequent molecular investigations.191
Genetic factors in the development of lung cancer
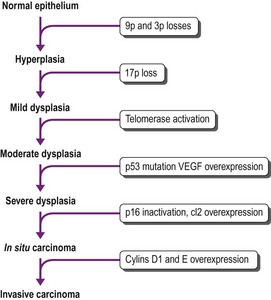
That the morphological changes described above are truly premalignant is supported by the demonstration that they show genetic abnormalities similar to those found in adjacent invasive cancer.192,193 They reflect a multistep accumulation of genetic abnormalities, the number of which gradually increases as dysplasia progresses (Fig. 12.1.6).128,194–196 The earliest molecular changes may be detected before any abnormality is evident at the microscopic level. Mutations involving a single gene are generally insufficient to cause cancer. Successive mutations affecting 10–20 different genes are required before malignant change is eventually established.197 Molecular techniques therefore enable the identification of premalignant change in tissues that may be normal histologically.198–202 Some of the earliest genetic changes involve loci on chromosomes 3p and 9p.203,204 Inactivation of the p17 gene is also an early event, whereas mutations involving the K-ras and p53 genes occur late in the sequence of genetic alterations.196,205 These genetic changes promote malignant transformation in a variety of ways, including enhanced cell growth by means of oncogene activation, inactivation of tumour suppressor genes, inhibition of apoptosis, altered DNA repair leading to cellular immortalization and the promotion of angiogenesis.196 Important growth factors that have been implicated in lung cancer are listed in Box 12.1.3. They are obviously important to the prognosis of lung cancer and are touched upon again under that heading on page 573.
Oncogene activation
Some of the oncogenes involved in lung cancer belong to the epidermal growth factor receptor family, EGFR1 (HER1), EGFR2 (HER2/neu, c-erbB2), HER3 and HER4. They act as transmembrane tyrosine kinase (TK) receptors, so promoting TKs, enzymes that regulate the activity of other proteins and which have been implicated in the development of many cancers. They are most commonly situated on chromosomes 19 and 21 and their presence predicts TK inhibitor response.206 They are targeted by the small-molecule TK inhibitors, such as gefitinib and erlotinib, and by monoclonal antibodies such as cetuximab and trastuzumab (Herceptin, which counters HER2).206–210 EGFR genes are especially overexpressed in papillary adenocarcinomas, particularly those arising in East Asian female non-smokers.210–212 Other EGFR mutations are associated with drug resistance. EGFR mutations occur early in tumour development with increased copy number being a late event.213
A structural homology between certain growth factor receptors and oncogenes probably underlies the autocrine activity evinced by certain tumours, in which the tumour secretes substances that induce self-proliferation. Gastrin-releasing peptide (human bombesin) is one of the best-known autocrine factors and is particularly involved in the remarkably rapid growth of small cell carcinomas.214 The gastrin-releasing peptide receptor gene appears to be activated by cigarette smoke and, being located on the X chromosome, is more strongly expressed in women than men,215 possibly contributing to the greater female susceptibility to tobacco-induced lung cancer noted above.4
Other oncogenes important in pulmonary carcinogenesis include members of the ras and myc gene families, especially K-ras and c-myc. Activating point mutations of ras genes result in an aberrant p21 membrane-associated protein that continuously transduces an inappropriate growth signal. K-ras is mutated in 30% of pulmonary adenocarcinomas,216 and particularly in smokers with mucinous adenocarcinomas.180,212,217 It is recorded in the precursor lesion atypical alveolar hyperplasia that is described above. K-ras mutation is therefore a good candidate for the early detection of lung cancer as it should be detectable in bronchioloalveolar cells shed in sputum or recovered by lavage. K-ras and EGFR mutations are mutually exclusive, the former being involved mainly in smokers’ tumours and the latter mainly in non-smokers’ adenocarcinomas.210 Furthermore, K-ras mutation is possibly responsible for resistance to cisplatin and the TK inhibitors referred to above. In adenocarcinoma, a pathway involving K-ras appears to be predominant in western smokers whereas the EGFR pathway appears to be more important in East Asian female non-smokers.210,211
Myc oncogenes encode for cell cycle-specific nuclear phosphoproteins and thereby control cell growth and differentiation. Their amplification accounts for myc protein accumulation in 20% of small cell carcinomas and 10% of non-small cell carcinomas. Myc gene amplification is found particularly in patients with recurrent tumour.218–220 Conversely, myc genes are seldom amplified in carcinomas from patients who have not undergone treatment and thus appear to be associated with tumour progression rather than initiation. Some drug regimens result in myc amplification more than others.
Fusion genes have long been recognised as being important in certain haematological malignancies and sarcomas but have not been thought to play a major role in epithelial tumours. Recently however the fusion gene EML4-ALK has been identified in non-small cell lung cancer as an alternative promoter of TK activity, thus opening up the possibility of further TK inhibitors being developed.221 The EML4-ALK gene has been found in about 5–10% of lung cancers, mainly adenocarcinomas in younger non-smokers.222–224 BRAF gene mutations are also seen in a minority of non-small cell lung cancers, again mainly adenocarcinomas.225,226 The Akt/mammalian (mTOR) pathway is frequently activated in human cancers and plays an important role in small cell lung cancer biology.227
Further genes involved in malignant change include the human homologue of the Drosophila neurogenic gene hASH1, which promotes the growth of small cell carcinoma, large cell neuroendocrine carcinoma and some atypical carcinoids,228 and members of the Forkhead box (Fox) family, dysregulation of which has been demonstrated in squamous cell carcinomas of the lung, particularly those of a more malignant nature.229
Tumour suppressor genes
In contrast to oncogenes, tumour suppressor genes are generally recessive and mutation of both alleles is therefore required for their inactivation. The first mutation results in heterozygosity, which has little effect because the mutant gene is recessive, whereas mutation of the other allele involves complete loss of the gene. The second mutation can be detected following suitable amplification, for example by the polymerase chain reaction, as a single band on gel electrophoresis, a change that is often termed ‘loss of heterozygosity’. Tumour suppressor genes (antioncogenes) are believed to play an important role in the pathogenesis of lung cancer because deletions or mutations of certain chromosomes are frequently found, notably chromosomes 3p, 13q and 17p.230–232 13q is the site of the retinoblastoma gene, 17p the p53 gene and 3p the FHIT (fragile histidine triad) antioncogene. 3p deletion is particularly common in small cell carcinoma and 13q and 17p deletion in non-small cell carcinoma.233–235 FHIT deletion is one of the earliest mutations and p53 deletion one of the last in the bronchial cancer gene sequence but loss of the FHIT antioncogene is relatively late in the atypical adenomatous hyperplasia–adenocarcinoma-in-situ–invasive peripheral adenocarcinoma sequence.168 Mutation of the EGFR is also increasingly found in the development of peripheral invasive adenocarcinomas from atypical adenomatous hyperplasia.169,236 Mutation of p53 is the commonest genetic alteration in human cancer and occurs in over half of lung cancers; p53 is normally responsible for apoptosis and abnormalities of this gene are thought to permit unchecked cell proliferation. The normal p53 protein product is generally undetectable due to a short half-life but mutations frequently extend this so that the protein accumulates and can be detected imunocytochemically. Accumulation of p53 protein product has been associated with an adverse prognosis in lung cancer and may provide a marker for early diagnosis of premalignant states.237
The retinoblastoma gene (Rb) is the main effector of G1 arrest, which is mediated by p53 and utilised in the repair of DNA damage. Rb protein expression is lost in 80% of small cell lung cancer and in 15% of non-small cell lung cancer, but is not found in preinvasive lesions.238 Inactivation of Rb is often due to methylation of p16 or overexpression of cyclin D1.
Bax and bcl-2 (B-cell lymphoma-2) are further genes involved in apoptosis, which is promoted by the former and blocked by the latter. The bcl-2 gene is overexpressed in preinvasive lesions, a variable number of non-small cell carcinomas of the lung,239–241 most small cell carcinomas and a variety of other lung tumours showing neuroendocrine differentiation, thereby permitting unchecked cell division.242,243 A high bcl-2:bax ratio generally accompanies mutation of the p53 tumour suppressor gene.243
Cellular immortalisation
Telomerase is an enzyme that adds nucleotide repeats to the ends (telomeres) of chromosomes to compensate for the nucleotide losses that occur with each round of DNA replication.244–247 Normal somatic cells show no telomerase activity and stop dividing when their telomeres are sufficiently shortened. Telomerase can be detected in most lung cancers and is frequently found in bronchial epithelium showing premalignant change. The indefinite proliferation of cancer cells probably owes much to their telomerase activity.
Vascular endothelium growth factor and other autocrine factors
Angiogenesis is an important process in the dissemination of invasive growths and the autocrine production of vascular endothelium growth factor by such lung tumours is therefore important in pulmonary carcinogenesis.248,249 Other autocrine factors secreted by certain lung tumours include epidermal growth factor (the genetic control of which is considered above), gastrin-releasing peptide, transferrin, insulin-like growth factor and endothelin (Table 12.1.4).214,250–254
Table 12.1.4 Molecular markers associated with lung cancer
| Gene mutation | Chromosome involved | Type of mutation |
|---|---|---|
| Proto-oncogene activation | ||
| K-ras, Ha-ras, N-ras | 12p | Point mutation |
| EGFR-2 (Her-2/neu, c-erb-B2) Ki-ras, Ha-ras, N-ras | 17q | Translocation |
| c-myc, L-myc, N-myc c-erb-B2 (Her-2/neu) (epidermal growth factor receptor gene) | 8 | Translocation |
| bcl-2 c-myc, L-myc, N-myc | 14–18 | Translocation |
| Tumour suppressor gene inactivation | ||
| FHIT (fragile histidine triad gene) | 3p | Deletion |
| Retinoblastoma gene | 13q | Point mutation |
| p53 | 17p | Deletion |
| Telomerase activation | ||
Screening for lung cancer
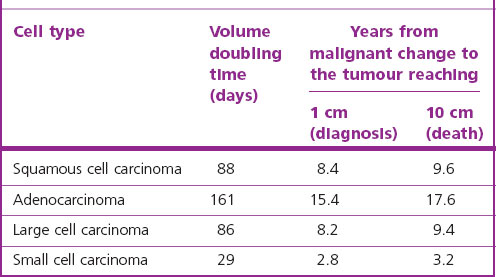
Lung cancer fulfils many of the criteria necessary for a successful screening programme. The condition is common, the population at risk is well known and premalignant changes can be detected cheaply (by sputum cytology). Unfortunately the premalignant changes cannot be easily eradicated: there is no bronchopulmonary equivalent of a uterine cone biopsy. Nor does screening for early invasive growths by a combination of sputum cytology and radiography appear to reduce mortality.255–257 The relative failure of surgery, radical radiotherapy and preventive screening programmes is partly explained by backward extrapolations of observed tumour size doubling times, which suggest that malignant change takes place years before a tumour is first detectable clinically (Table 12.1.5).258 Nevertheless, sparked by the advances in molecular pathology described above and refinements in high-resolution computed tomography, there has been a renewal of interest in early detection.255 For example, it appears that p53 immunohistochemistry may assist in the identification of preneoplastic lesions,192 the detection of p16INK4a inactivation in sputum may identify smokers at increased risk of developing lung cancer,259,260 and low-dose computed tomography (CT) screening trials are proving more sensitive in detecting small peripheral tumours.261–266 Wedge resection rather than lobectomy is undertaken for these small asymptomatic lesions, with good results, particularly for those showing ground-glass opacification, which is characteristic of adenocarcinoma-in-situ and minimally invasive adenocarcinoma of lepidic pattern.267–270 Nevertheless, evidence-based reviews do not support general CT screening,256,257 possibly because it identifies many unimportant abnormalities and leads to unnecessary surgery (see asymptomatic solitary pulmonary nodule, below).
Site of origin and spread
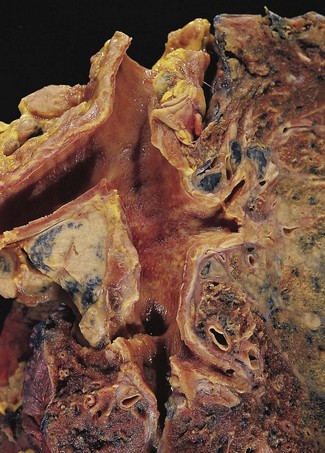
As might be expected from the relative size of the two lungs, slightly more carcinomas arise on the right than the left. There is also a slight preponderance in the upper lobes. Historically, most carcinomas arose in large central bronchi (Fig. 12.1.7) but increasing numbers now develop in the periphery of the lung so that whereas the ratio of central to peripheral tumours was formerly 2:1, it now approaches equality.271–274 This change has been accompanied by one involving the histological types of bronchopulmonary carcinoma, particularly squamous cell carcinoma (see Fig. 12.1.18, p. 550).
Within the conductive airways, carcinoma arises particularly at bifurcations where inhaled particles tend to impinge and mucociliary clearance is delayed (Fig. 12.1.8 and see Fig. 1.18, p. 13)275–277 It is probably because the trachea lacks side branches that primary carcinoma of the trachea is rare.278 The few tumours that arise in the trachea are generally of salivary gland type or carcinoids.279
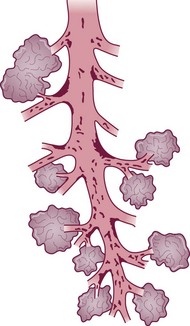
(Reproduced from Iravani and van As (1972)275 with permission of the editor of Journal of Pathology.)
Dissemination of the tumour is chiefly along lymphatic routes, at least initially. Blood spread usually follows. Metastases in hilar and mediastinal lymph nodes, and perhaps beyond, have often developed by the time symptoms first cause the patient to seek attention. Retrograde spread within the lung may lead to widespread lymphatic permeation, resulting in so-called lymphangitis carcinomatosa, which is described in Chapter 12.6, as are other routes of tumour spread within the lung. Further tumour growth and dissemination give rise to a variety of clinical effects, which will now be described.
Clinical effects
An asymptomatic solitary pulmonary nodule (‘coin lesion’)
The traditional management of an asymptomatic patient found to have a solitary, well-circumscribed pulmonary opacity has been based on the supposition that the lesion is malignant until proved otherwise. However, improvements in imaging have led to the realisation that small asymptomatic nodules are very common. Up to 51% of smokers older than 49 years have pulmonary nodules and the great majority of these prove to be benign (99% of those no greater than 4 mm diameter). It is therefore now recommended that nodules measuring 8 mm or less should be merely kept under surveillance.280 In nodules larger than 8 mm, central calcification and a slow growth rate suggest that the lesion is benign whereas fine linear strands radiating from the edge of the nodule, cavitation and air bronchograms favour the converse, but these features are not wholly reliable.281 Not infrequently all investigations short of thoracotomy fail to provide a diagnosis and the lesion is biopsied or resected. While it may well prove to be fungal in the USA, an echinococcal cyst in the Orient, and tuberculosis worldwide, a malignant tumour is often the cause (71% in a New Zealand series).282 Metastases account for about 20% of such malignancies but this figure rises to nearly 50% in patients with an extrapulmonary cancer.283
Local effects
Central tumours are prone to obstruct the larger airways (Fig. 12.1.9), leading to pulmonary collapse, infection and ‘endogenous lipid pneumonia’ (see p. 315). This results in cough, dyspnoea, pain and fever. Haemoptysis caused by ulceration of the tumour is another common symptom but is not usually severe. However, many peripheral tumours reach a substantial size and metastasise before causing any symptoms at all. Cavitation is often evident radiologically, particularly with squamous cell carcinomas, whilst the radiographic features of mucinous adenocarcinoma of predominantly lepidic pattern often simulate those of pneumonic consolidation. Patients being treated for pneumonia should therefore be observed until there is complete roentgenographic resolution. Any pneumonia that does not resolve within 4 weeks should be evaluated further to exclude carcinoma. The expectoration of large amounts of watery or mucoid sputum (bronchorrhoea) is occasionally encountered in patients harbouring adenocarcinoma-in-situ or mucinous adenocarcinomas of predominantly lepidic pattern.
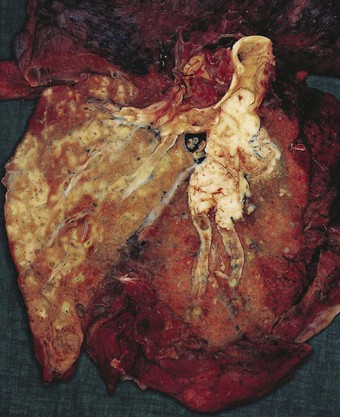
Intrathoracic spread
Direct spread may cause pleural or chest wall pain whilst either pleural involvement or poststenotic bronchopneumonia may cause pleural effusion, which if neoplastic is characteristically haemorrhagic (Fig. 12.1.10).
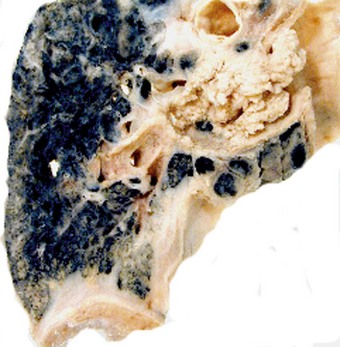
Figure 12.1.10 Carcinoma of the bronchus with extensive pulmonary and pleural extension.
(Courtesy of W Edwards, Gordon Museum, Guy’s Hospital, London, UK.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


