Chapter 24 Tumors of the surface epithelium
Epidermal nevi
Clinical features
Lesions, which may be present at birth or develop during childhood, are usually yellowish-brown warty papules or plaques with irregular margins. They commonly affect the trunk or limbs and vary from trivial small lesions to very extensive areas of involvement that may cause the patient great cosmetic embarrassment (Fig. 24.1). The sex incidence is equal and there is no racial predilection. Familial cases may rarely be seen and are transmitted in an autosomal dominant fashion.4
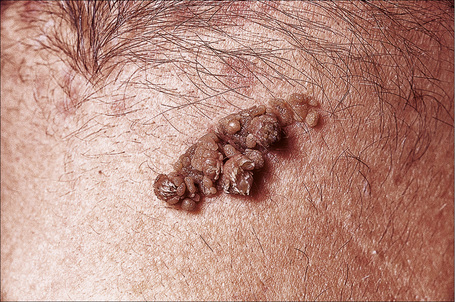
Fig. 24.1 Epidermal nevus; this fairly typical lesion presented on the chest of a young male.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Nevus verrucosus refers to solitary or multiple localized lesions, nevus unius lateris refers to a more severe unilateral linear distribution, and ichthyosis hystrix refers to the most extreme example with a bilateral or generalized distribution (Figs 24.2, 24.3). Rare but important complications are development of basal cell and squamous cell carcinoma in addition to keratoacanthoma, eccrine syringofibroadenoma, syringocystadenoma papilliferum and clear cell acanthoma.5–15 Unusual presentations include onset in adulthood, involvement of the maxilla, the palm or oral mucosa as well as a bilateral symmetric distribution.16–21 Association with a number of diseases and syndromes have been reported including keratitis-ichthyosis-deafness (KID) syndrome, Gardner’s syndrome, Rubinstein-Taybi syndrome, precocious puberty, hypophosphatemic vitamin D-resistant rickets, digital constrictions, divided fingers, localized cranial defects, hemimegalencephaly, temporal lobe enlargement, vascular anomalies and malformations, renal artery stenosis, polyostotic fibrous dysplasia, choristomas, hypermelanosis and chronic hyponatremia, segmental hypermelanosis, trichilemmal and proliferating trichilemmal cysts, CNS lipomas, mandibular ameloblastoma, chondroblastoma, rhabdomyosarcoma, transitional cell carcinoma multiple apocrine adenoma, and malignant eccrine poroma.22–58 Epidermal nevi may also present together with nevus sebaceus, woolly hair nevus, and nevus comedonicus.13,59–64 They may also rarely be complicated by a dermatitis such as psoriasis.63
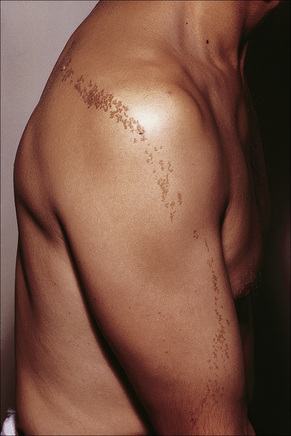
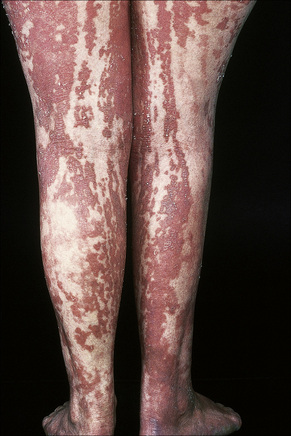
Fig. 24.3 Ichthyosis hystrix: in this variant there is extensive and bilateral involvement.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Patients with the epidermal nevus syndrome show a variety of systemic manifestations. They also have an increased incidence of café-au-lait macules, congenital hypopigmented macules, congenital as well as Spitz melanocytic nevi, and a range of vascular malformations. Visceral features particularly affect the skeletal, ocular, and central nervous systems.1,2,5,65–67 These include kyphoscoliosis, bone cysts, genu valgum, skull abnormalities, limb reduction defects, strabismus, mental retardation, delayed motor milestones, cranial nerve palsies and nonfebrile seizures.1,2,5,68 Patients with the epidermal nevus syndrome may have an increased incidence of systemic malignancy.1 More recent studies suggest that the epidermal nevus syndrome is a heterogeneous group of disorders that can be separated by clinical and pathological as well as genetic criteria.69,70 At least six distinct syndromic entities are recognized, each associated with distinct epithelial nevi: 51,52,70–74
Furthermore, mosaic chromosome trisomy 6 has recently been documented in a patient with a linear epidermal nevus without systemic manifestation, and a mosaic FGFR3 mutation has been detected in a patient with epidermal nevus syndrome with cerebral involvement.75,76
Schimmelpenning syndrome
Schimmelpenning syndrome (Schimmelpenning-Feuerstein-Mims syndrome, organoid nevus phakomatosis) is characterized by unilateral sebaceus nevus of the head and neck in combination with cerebral, ocular, cardiac, vascular, and skeletal abnormalities.69,70,77–85 It is believed to represent a lethal autosomal inherited condition with survival dependent upon a mosaic state.69 A dominant variant presenting in one of two monozygotic twins has recently been reported.79 Neurological abnormalities include mental retardation, convulsions, hemiparesis, cranial asymmetry, and hydrocephalus. Oral involvement has also been documented. Patients may present with gingival papillomata, hemihypertrophy of the tongue, bone cysts, aplasia of teeth, and hypoplastic or absent enamel as well as pigmented malformed teeth, multiple odontomas, bilateral maxillary fibro-osseous lesions, recurrent central giant cell granulomas of the jaw, and adenomatoid odontogenic tumor.86 Ocular manifestations take the form of microphthalmos, eyelid coloboma, nystagmus, ptosis, dermoids, and teratomas of the conjunctiva.58,62 Skeletal abnormalities include skull asymmetry, exostoses, and scoliosis.62
Nevus comedonicus syndrome
The nevus comedonicus syndrome consists of nevus comedonicus, cataract, and skeletal defects.
Pigmented hairy epidermal nevus syndrome
The pigmented hairy epidermal nevus syndrome comprises Becker’s nevus in association with ipsilateral hypoplasia of the breast and other cutaneous as well as skeletal anomalies.70
Proteus syndrome
Proteus syndrome is an exceedingly rare hamartomatous condition which can be very disfiguring and is associated with an increased risk of malignancy. It is sporadic and is thought to result from mosaicism for a mutation that is lethal in the nonmosaic state.87,88 Males are more frequently affected than females and the syndrome is associated with an increased risk of premature death.89 A subset has been linked to PTEN mutations, thereby displaying genetic in addition to clinical overlap with two other hamartomatous conditions: Cowden’s syndrome and Bannayan-Riley-Ruvalcaba syndrome.88,90–94 Recent mutational analyses of patients with ‘bona fide’ Proteus syndrome, however, failed to detect mutation in the PTEN gene and the contribution of PTEN mutations remains an issue of ongoing debate.95,96 Proteus syndrome is a complex disorder comprising malformations and overgrowth of multiple tissues in addition to epidermal nevi. The latter are flat, soft, and of nonorganoid type and are present in about two-thirds of cases.69,97–100 They are associated with other cutaneous anomalies such as connective tissue nevi, cystic lymphangiomas, hemangiomas, lipomas, and fibromas.101–104 Cerebriform hyperplasia of the plantar connective tissue (moccasin lesion) is a characteristic finding.69,103 A hallmark of Proteus syndrome is asymmetrical hypertrophy affecting the face, limbs, and trunk including macrocephaly and hemihypertrophy of the body.87,88 Macrodactyly is thought to be characteristic.98
Other noncutaneous manifestations include bony exostoses, kyphoscoliosis, spinal canal stenosis, hydrocephalus, hemimegancephaly, CNS cysts, seizures, epibulbar tumors, enlargement of the eye, cataract and strabismus, paraovarian endometrioid cystic tumors, genitourinary abnormalities, and nephrogenic diabetes insipidus.98,105–107 Patients are at increased risk for a number of tumors including testicular papillary adenocarcinoma, ovarian cystadenoma, meningioma, parotid monomorphic adenoma, astrocytoma, optic nerve tumor, leiomyoma, and endometrial carcinoma.83,95
Recently, two patients from families with Cowden’s syndrome have been described with a unique phenotype related to bi-allelic inactivation of PTEN. These patients showed classic features of Cowden’s syndrome in addition to segmental overgrowth, lipomatosis, arteriovenous malformation as well as linear epidermal nevus for which the term SOLAMEN syndrome has been proposed.108
CHILD syndrome
CHILD syndrome is an X-linked dominant disorder almost exclusively affecting females and is characterized by a unique epidermal nevus involving one side of the body in addition to ipsilateral defects of the limbs and internal organs.109 Mutations in the NSDHL (NADPH steroid dehydrogenase-like protein) gene, which is located on Xq28 and is involved in the cholesterol synthesis pathway, have been identified in this syndrome.110–114 The CHILD nevus is a unilateral, circumscribed, inflammatory, ichthyosiform nevus typically presenting at birth. It is erythematous and covered by yellow, waxy scales.115,116 Frequently, the body folds are affected. It may be linear and follow the lines of Blaschko. Disease presentation can be mild and familial, affecting multiple generations.117
Histologically, the cutaneous manifestations are characterized by psoriasiform epidermal hyperplasia, and features reminiscent of verruciform xanthoma may be present. Ultrastructural data suggest that the formation of the cutaneous lesion may be due to abnormal lipid metabolism.118 Although CHILD nevus has a distinctive presentation there is considerable overlap with inflammatory linear verrucous nevus (ILVEN).115,116 Other clinical findings in CHILD syndrome include ipsilateral hypoplasia or aplasia of limbs and other skeletal structures. Neurological defects may include hypoplasia of a cerebral hemisphere or cranial nerve but mental development is typically unimpaired. Cardiovascular, renal, and pulmonary anomalies may be present.70 Development of a cutaneous squamous cell carcinoma has recently been reported.119
Phakomatosis pigmentokeratotica
This recently described syndrome is characterized by the presence of a nevus sebaceus following the lines of Blaschko in addition to a speckled lentiginous nevus (nevus spilus) typically in a segmental distribution with a checkerboard pattern.74,120–123 Extracutaneous manifestations are typically but not invariably present.124,125 They comprise predominantly skeletal, neurological, and ocular abnormalities and the most consistent extracutaneous finding is hemiatrophy.74,120–123 Neurological findings include hyperpathia, dysesthesia, hyperhidrosis, seizures, deafness, ptosis, strabismus, and mild mental retardation.74,120–123,126 Other associations include hypophosphatemic vitamin D-resistant rickets, hemihypertrophy, linear connective tissue nevus, juvenile onset hypertension, aortic stenosis, dermoid cysts, pheochromocytoma, and rhabdomyosarcoma.127–132 A melanoma arising in a nevus spilus has been documented and the organoid nevus may be complicated by the development of multiple basal cell carcinomas.128,133,134
Histological features
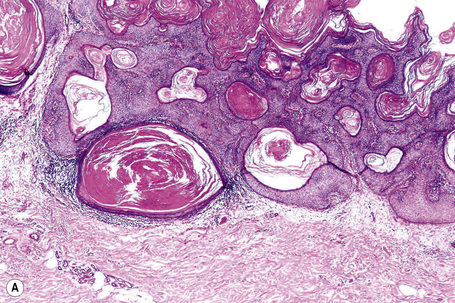
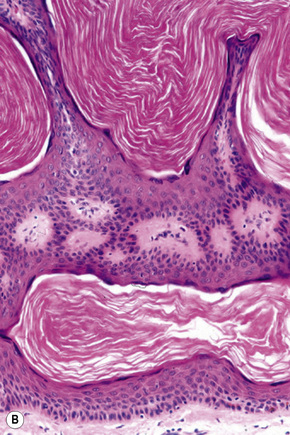
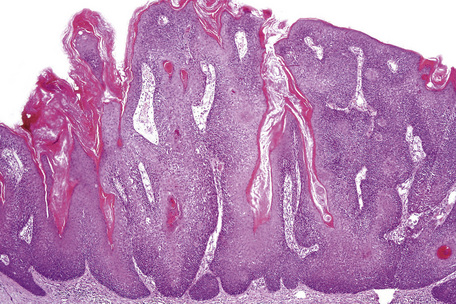
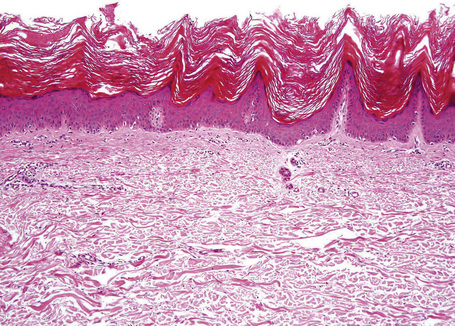
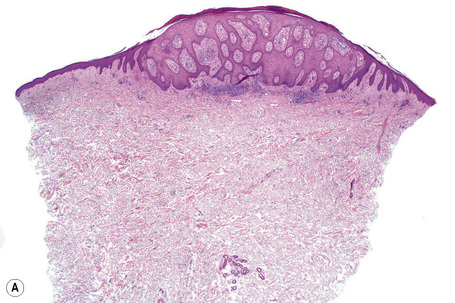
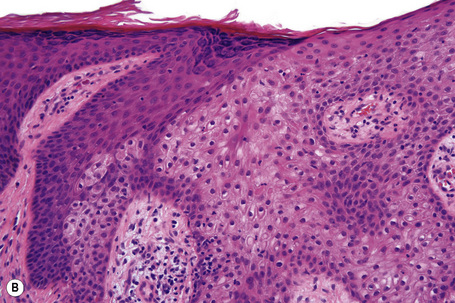
Epidermal nevi represent developmental abnormalities limited to proliferation of the epidermis, sometimes accompanied by anomalous terminal differentiation. Although they are related to sebaceus (organoid) nevi and frequently coexist, it is convenient to consider them separately. Commonly, they show the features of a sharply demarcated simple squamous cell papilloma (Fig. 24.4). They therefore manifest hyperkeratosis, papillomatosis, acanthosis, and elongation of the rete ridges (Fig. 24.5).135 The histological features are often subtle and occasionally resemble acanthosis nigricans or a seborrheic keratosis.136 Rarely, the lesions show focal acantholytic dyskeratosis, acrokeratosis verruciformis-like features, cornoid lamellae formation, epidermolytic hyperkeratosis, an associated lichenoid tissue reaction or cutaneous horn formation.137–142 Acantholytic dyskeratosis in epidermal nevi is thought by some authors to represent zosteriform Darier’s disease.143–145 The presence of epidermolytic hyperkeratosis may be indicative of mosaicism for bullous ichthyosiform erythroderma as further supported by the identification of keratin 1 and 10 mutations.146–149
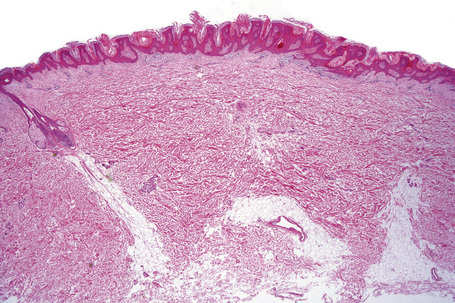
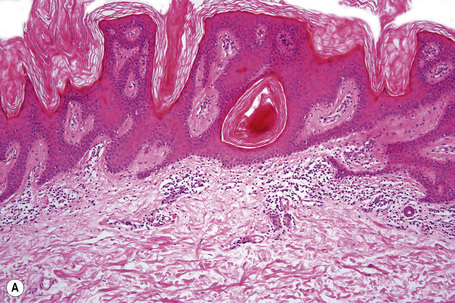
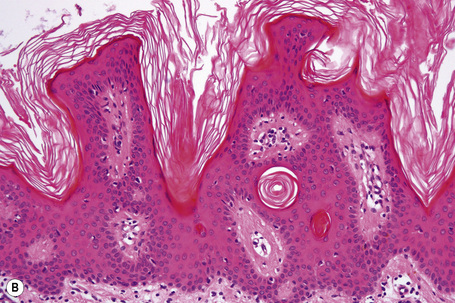
Epidermal nevi in Proteus syndrome are characterized by hyperorthokeratosis, acanthosis and papillomatosis in the absence of adnexal hyperplasia or epidermolytic hyperkeratosis.69
The CHILD nevus is characterized histologically by marked acanthosis with overlying parakeratosis admixed with areas of orthohyperkeratosis. Intraepidermal neutrophils, also forming aggregates similar to Munro’s abscesses, are sometimes present. The dermis shows a lymphohistiocytic infiltrate. Dermal papilla may be filled with foamy histiocytes reminiscent of verruciform xanthoma.116,150
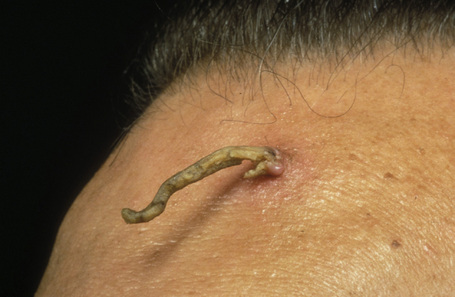
Cornu cutaneum
Cornu cutaneum (cutaneous horn) is a clinical diagnosis based upon the presence of a large protuberant mass of keratin (Fig. 24.6).1–7 It may complicate a variety of conditions and accurate diagnosis depends upon histological examination of tissue from the base of the lesion. Cutaneous horns may complicate solar keratosis, viral wart, seborrheic keratosis, squamous cell carcinoma, keratoacanthoma, lichenoid keratosis, and basal cell carcinoma.3
Acanthoma fissuratum
Clinical features
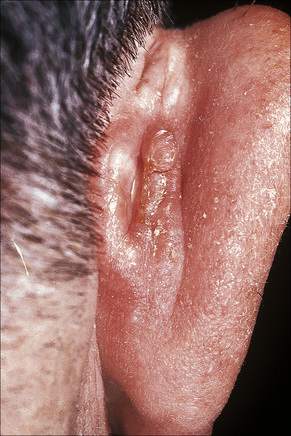
Acanthoma fissuratum (spectacle frame acanthoma) is a benign epidermal ‘tumor’ which develops as a result of chronic irritation from spectacles.1–5 It presents as an erythematous or flesh-colored, sometimes tender, nodule situated on the bridge of the nose or behind the ears (Fig. 24.7).1–5 Typically, the center of the lesion shows a linear groove. The importance of this entity is that it is clinically frequently misdiagnosed as a basal cell or squamous cell carcinoma. An analogous lesion termed granuloma (epulis) fissuratum has been described in the mouth as a result of poorly fitting dentures.6
Histological features
The histopathology is not specific, diagnosis being dependent upon knowledge of the clinical history. Its features usually include acanthosis (sometimes amounting to pseudoepitheliomatous hyperplasia) with hyperkeratosis and patchy parakeratosis, particularly at the edges of the fissure (Fig. 24.8).7 The underlying dermis is edematous, fibrosed, and hyalinized. A chronic inflammatory cell infiltrate of variable severity is commonly present.
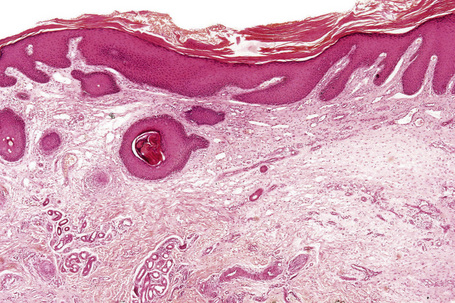
Seborrheic keratosis
Clinical features
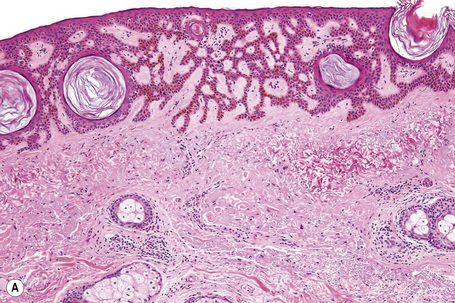
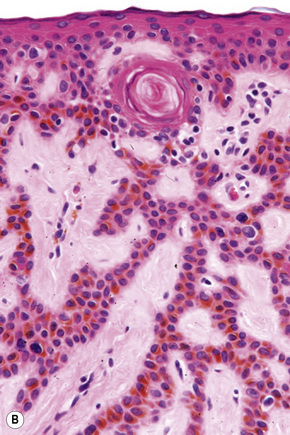
Seborrheic keratoses are very common lesions, developing in the middle aged and elderly. They are frequently numerous and appear as sharply delineated, round or oval, flesh-colored or brown-black warty plaques with a rather greasy texture (Figs 24.9, 24.10).1 Sometimes they are dome-shaped with a smooth surface and occasionally they may show an inflammatory halo or eczema-like features (Meyerson’s phenomenon).2–5 Although they may be found anywhere on the body (except for the palms and soles), they are particularly common on the face, chest, and back.1 The conjunctiva may also be involved.6
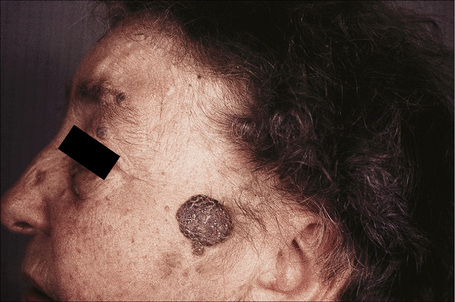
Fig. 24.9 Seborrheic keratosis: multiple lesions on an elderly lady.
By courtesy of the late M. Beare, MD, Royal Victoria Hospital, Belfast, N. Ireland.
Other unusual clinical presentations include distribution along skin cleavages,7 involvement of the areola8,9 or the back of elderly patients (raindrop seborrheic keratosis) as well as a distribution along the lines of Blaschko.10,11 Occasionally, deeply pigmented or traumatized lesions are mistaken clinically for melanoma. The irritated seborrheic keratosis (inverted follicular keratosis) presents as a small warty papulonodule (Fig. 24.11). It commonly affects middle-aged or elderly males and shows a predilection for the face.12 Multiple inverted follicular keratoses have been reported as a presenting sign in a patient with Cowden’s syndrome.13
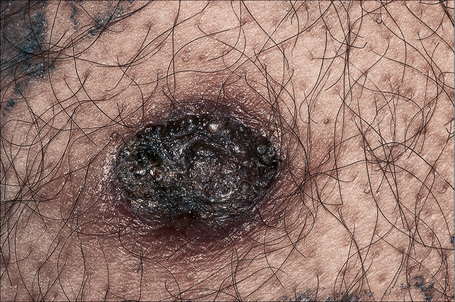
Sudden onset of numerous seborrheic keratoses (Leser-Trélat sign) has been reported in association with internal malignancy, most commonly adenocarcinoma of the stomach. Other tumors which may be present include lymphoma (particularly mucosis fungoides and Sézary syndrome), leukemia, bronchial and breast carcinoma, cholangiocarcinoma, carcinoma of the ampulla of Vater, the pancreas and the esophagus, adenocarcinoma of the rectum, renal cell carcinoma, transitional cell carcinoma, and anaplastic ependymoma (Fig. 24.12).14–31 Leser-Trélat sign has also been described after treatment with chemotherapy as well as in association with a benign Leydig cell tumor.32,33 The relationship of seborrheic keratosis and malignancy, however, has been questioned.34,35 Although neoplasia and seborrheic warts are both common in the elderly, we believe that a sudden eruption of the latter should still warrant careful investigation. Spontaneous regression of multiple seborrheic keratoses has also been reported in association with malignancy.36 Multiple eruptive seborrheic keratoses have been observed in a background of erythroderma, at a postoperative site as well as with leprosy.37–40
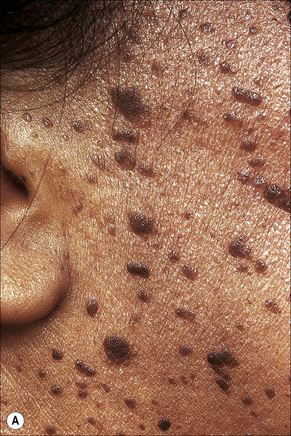
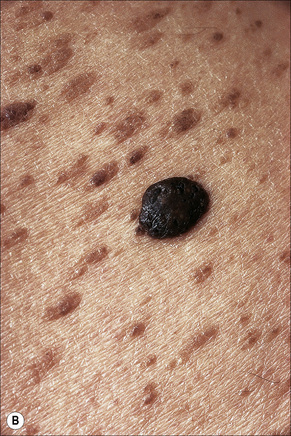
Histological features
This benign epidermal tumor has recently been shown to be monoclonal in nature and to represent a neoplasm rather than epidermal hyperplasia.41 It is characterized by proliferation of basaloid cells with a variable degree of squamoid differentiation.1 There is a range of histopathological patterns, including the hyperkeratotic (papillomatous) and adenoid variants, acanthotic keratosis, and irritated seborrheic keratosis (inverted follicular keratosis).42
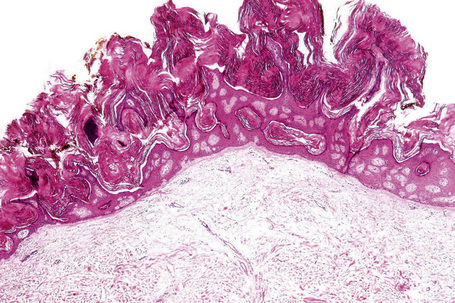
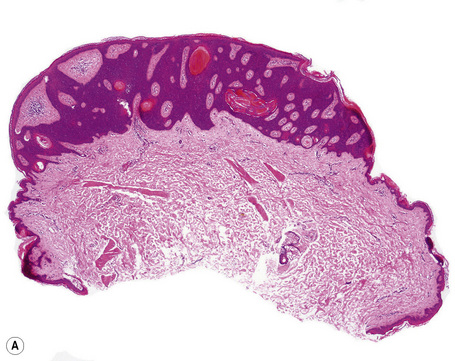
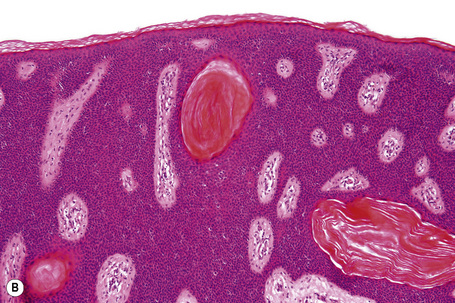
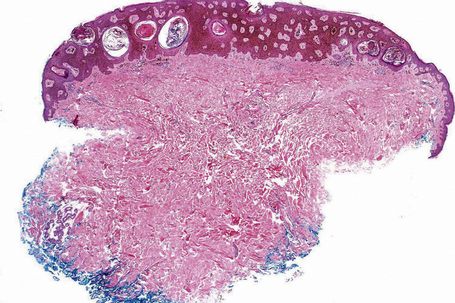
Many seborrheic keratoses show a mixture of the patterns described above. Occasionally, intraepithelial nesting gives rise to the Borst-Jadassohn appearance (so-called clonal seborrheic keratosis) and some variants barely protrude above the adjacent epidermis (flat seborrheic keratosis) (Figs 24.19–24.21). Rare patterns include the adamantinoid variant characterized by intercellular mucin and pseudorosette formation due to a radial arrangement of basal keratinocytes around central small empty spaces.45 The presence of numerous basal cells with abundant clear cytoplasm is a rare finding and may lead to an erroneous diagnosis of melanoma in situ within a seborrheic keratosis on H&E-stained sections.46 By immunohistochemistry, these basal clear cells are, however, positive for cytokeratins and negative for markers of melanocytic differentiation, confirming their epithelial nature.46 The etiology of marked clear cell change is unclear but appears unrelated to glycogen deposition. A desmoplastic variant is characterized by nests and strands of bland-appearing basaloid epithelium within a dense desmoplastic stromal response. Analogous to desmoplastic trichilemmoma, the tumor shows an exophytic growth pattern, is well-demarcated, and lacks cytological atypia.47 Stromal amyloid deposition may occasionally be evident, and an accumulation of xanthomatized histiocytes within dermal papillae reminiscent of verruciform xanthoma has been reported.41,48
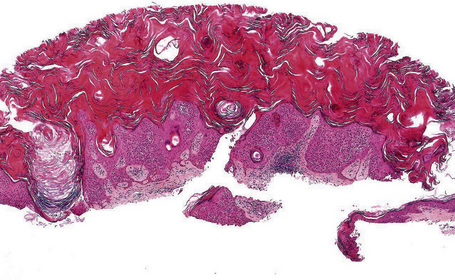
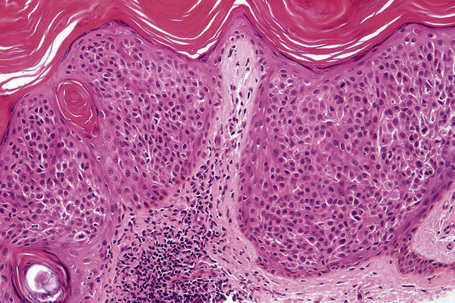
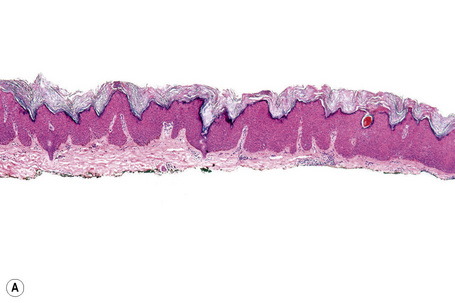
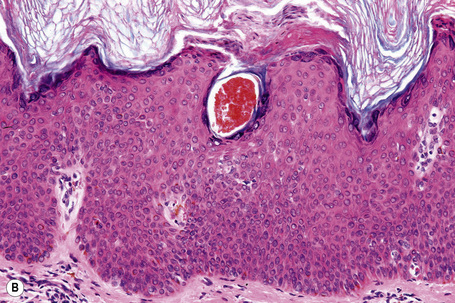
Fig. 24.21 (A, B) Flat seborrheic keratosis: this variant may clinically be mistaken for Bowen’s disease.
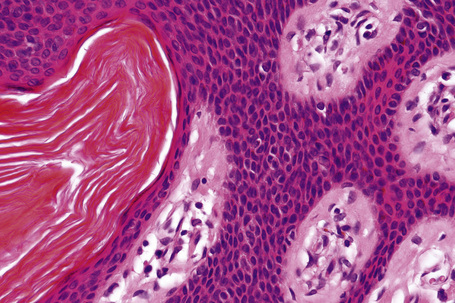
Seborrheic keratosis is sometimes characterized by an intradermal or ‘inverted’ type of proliferation known as inverted follicular keratosis (Fig. 24.22).12 In this variant characteristic whorls of maturing squamous epithelium (squamous eddies) are particularly evident (Figs 24.23, 24.24). These tumors are usually seen arising close to a hair follicle ostium and squamous eddies have been shown to be related to follicular units.49 Mitotic figures are sometimes conspicuous, but are invariably normal.12 Some of the lesions contain large numbers of horn cysts and are frequently accompanied by an intense chronic inflammatory cell infiltrate, hence its alternative designation, ‘irritated seborrheic keratosis’. Conspicuous apoptosis is sometimes present in areas of squamous differentiation.50
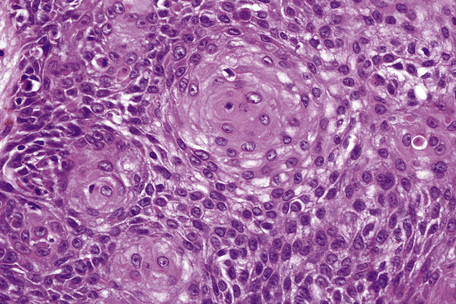
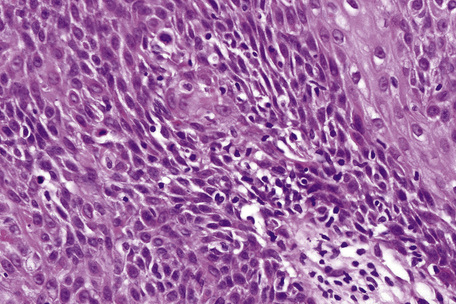
Acantholysis is not uncommon in seborrheic keratoses, particularly in the ‘irritated’ variant.51,52
Malignant change within a seborrheic keratosis is rare, but has occasionally been documented.53–55 Usually this represents an in situ change, but basal cell carcinoma and squamous cell carcinoma have also been reported.56,57 More frequently (in about 5% of tumors) neoplasms arise in association with seborrheic keratoses, sometimes referred to as collision tumors.58 The most frequent association is with superficial basal cell carcinoma but squamous cell carcinoma and melanoma have also been reported.56,59–66 Unusual combined tumors that have very occasionally been documented include cutaneous ganglioneuroma, melanocytic nevi, sebaceoma, eccrine poroma, and trichilemmoma in addition to adenocarcinoma.67–74
In contrast to nongenital lesions, seborrheic keratoses arising on genital skin are frequently (in approximately 70%) related to HPV.75–78 These HPV-positive lesions are probably better regarded as condyloma acuminatum rather than true seborrheic keratoses.79
Dermatosis papulosa nigra
Clinical features
Dermatosis papulosa nigra is an extremely common condition in which adult Afro-Caribbeans develop multiple, small, darkly pigmented papules, predominantly on the face, particularly the cheek.1 The neck, chest, and upper back may also be involved.2–5 There is a female preponderance of 2:1, and a family predisposition has been identified in the majority of patients.3,6 Very rarely, children may be affected and exceptionally the disorder presents in the white races.4,5 An eruptive presentation has been reported in a patient with a colonic adenocarcinoma analogous to eruptive seborrheic keratoses associated with internal malignancy (Leser-Trélat sign).7
Histological features
The lesions are indistinguishable from seborrheic keratoses (Fig. 24.25). There is hyperkeratosis, acanthosis with a reticulated pattern, and ‘horn cyst’ formation.2 The epithelium shows abundant melanin pigmentation. A chronic inflammatory cell infiltrate may be present in the superficial dermis.
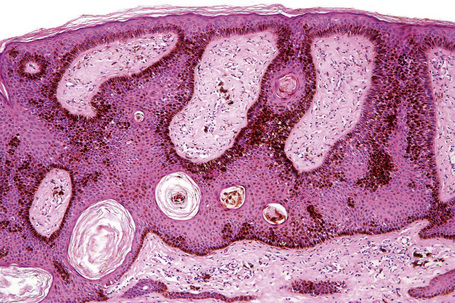
Large cell acanthoma
Clinical features
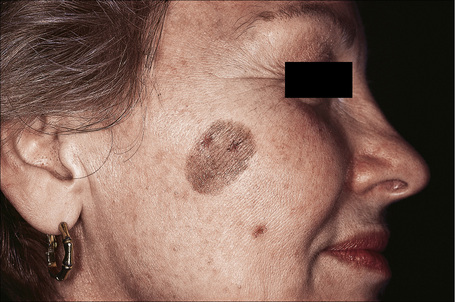
Large cell acanthoma presents as a discrete scaly papule or plaque up to 1 cm or more in diameter and is most commonly found on the head, arms, trunk, and lower limbs in decreasing order of frequency (Fig. 24.26).1–4 Lesions, which are sharply demarcated, are often single, but multiple acanthomas have occasionally been documented.1,5 The papules are commonly hyperpigmented or flesh colored, although achromic lesions have also been described.6 The sex incidence is equal and patients are usually elderly. Large cell acanthoma does not therefore have distinctive clinical features, most examples being submitted as seborrheic keratoses, actinic keratoses or actinic lentigines.2
Pathogenesis and histological features
The precise nature of this curious lesion is unknown. It has been variably described as a distinct entity, a variant of actinic keratosis or Bowen’s disease, a form of stucco keratosis or a subtype of solar lentigo.1,2 The recent finding of aneuploidy in this clinically benign acanthoma suggests that it merits separate classification.3,7,8 Human papillomavirus (HPV) type 6 has been identified from lesional skin in one patient with multiple disseminated large cell acanthomata.9
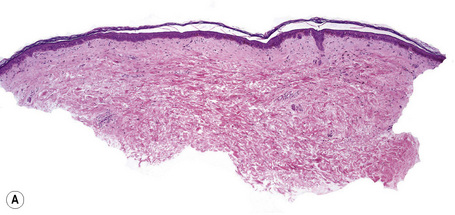
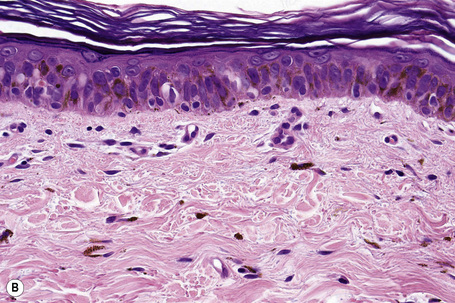
Histologically, large cell acanthoma is composed of keratinocytes measuring about twice normal size (Fig. 24.27).10,11 Hyperkeratosis is usual and the acanthotic epithelium shows bulbous epidermal ridges. Keratinocytes contain enlarged and rounded nuclei and the cytoplasm is abundant. Cytological atypia is not a feature and mitotic activity is limited to basal keratinocytes. Lesions may be hyperpigmented, normally pigmented or achromic. Verrucous variants reminiscent of stucco keratoses have also been described.1
Stucco keratoses
Clinical features
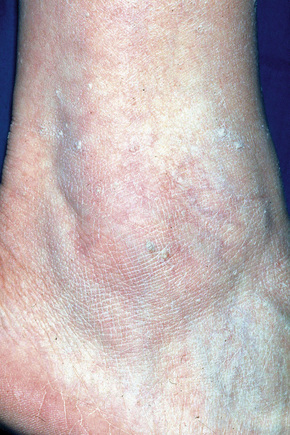
Stucco keratoses are warty lesions found on the extremities of elderly people and are more common in males than in females.1–3 They are small (1–2 mm), sharply defined, round or oval verrucous papules of gray or brown coloration, commonly located around the foot, ankle, and dorsum of the hand (Fig. 24.28).2,3 The extensor surface of the forearm is also often affected. The lesions are typically numerous, often in excess of a hundred. The papules appear stuck to the underlying skin, but can be easily scratched off, leaving an intact undersurface with a scaly collarette.2 Xerosis is often present and patients may also show scattered seborrheic keratoses.
Pathogenesis and histological features
The etiology and pathogenesis of this disorder is still uncertain. Ultrastructural studies have shown normal epidermal differentiation without evidence of intracellular viral particles.4 However, a single study of one patient with disseminated eruptive lesions demonstrated human papillomavirus DNA.5
The lesions are typified by dense orthokeratosis and ‘peaked’ or church-spire acanthosis (Fig. 24.29).2,3 Basaloid cell proliferation and horn cyst formation as seen in a seborrheic keratosis are characteristically absent.3 There are no significant inflammatory changes.
Intraepidermal epithelioma of Borst-Jadassohn
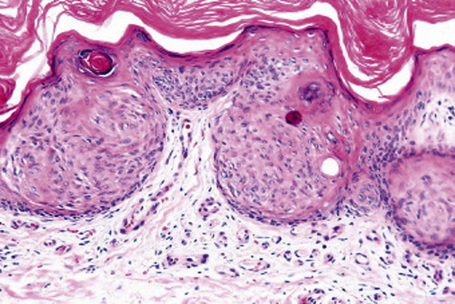
The Borst-Jadassohn epithelioma refers to a histopathological appearance rather than a precise clinicopathological entity.1 It had initially been regarded as a distinctive clinicopathological entity.2 The histological findings, however, are seen in a number of lesions of different etiology such as Bowen’s disease, actinic keratosis, hidracanthoma simplex, and seborrheic keratosis and it is therefore preferable to regard this as a histological pattern rather than a distinct entity.3,4 Histologically, it is characterized by nests of neoplastic cells situated within and surrounded by normal keratinocytes (Fig. 24.30).
Porokeratoma
Clinical features
Porokeratoma is an only recently described entity characterized by histological features of porokeratosis presenting as solitary lesions. It shows a marked male predominance and affects adults with a mean age of 57 years.1 The clinical presentation is of a hyperkeratotic plaque or nodule which may appear verrucous. A wide range of anatomic sites is affected but there is a predilection for distal extremities.1 There is no underlying or associated porokeratosis and behavior is benign.
Histological features
Histologically, porokeratoma is well circumscribed and characterized by marked epidermal hyperplasia and papillomatosis showing prominent distinct or broad and confluent cornoid lamella formation with dyskeratosis and loss of the granular cell layer.1 There is an abrupt transition with areas of adjacent orthohyperkeratosis. The dermis shows a non-specific chronic inflammatory infiltrate and mild vascular dilatation.
Psoriasiform keratosis
Clinical features
Psoriasiform keratosis typically presents as solitary and occasionally multiple erythematous scaly papules or plaques measuring between 0.5 and 3 cm.1,2 The anatomic distribution is wide and there is a predilection for the extremities. Adults are affected, with a mean age at presentation of 66 years.1,2 The gender distribution is roughly equal and there is no history of psoriasis. Behavior is benign.
Histological features
The histological findings are at least somewhat reminiscent of psoriasis. There is sharp lesional circumscription and marked irregular, verrucous epidermal acanthosis with overlying hyperparakeratosis.1,2 Focal to confluent mounds of parakeratosis contain collections of neutrophils and there is an absence or diminution of the granular cell layer in these areas.1,2 Prominent and dilated vessels are present within the papillary dermis, accompanied by a chronic inflammatory infiltrate predominantly containing lymphocytes. A fungal etiology is excluded by PAS staining.
Granular parakeratotic acanthoma
Clinical findings
This solitary keratosis presents in adulthood with a median age of 59 years. Trunk and extremities are mainly affected and, based on the limited information available, there is a slight female bias.1
Histological features
The histological hallmark is the presence of keratinocytes showing conspicuous granular parakeratosis as observed in granular parakeratosis.1 The lesion is otherwise circumscribed, with an endophytic growth pattern reminiscent of the adenoid pattern in seborrheic keratosis showing infundibular pseudocyst formation.1 There is a florid accompanying chronic inflammatory infiltrate within the superficial dermis and lichenoid features may be present. Eosinophils and histiocytes are admixed.
Clear cell acanthoma
Clinical features
Clear cell acanthoma (Degos) is an uncommon, usually solitary, tumor occurring in the middle aged or elderly but which may rarely present in younger patients.1,2 Occasional multiple3–5 or disseminated eruptive6–8 variants have been described. It is most commonly found on the lower limbs and presents as a circumscribed pink to bright red or brown oval-shaped papule or nodule measuring 1–4 cm in diameter (Fig. 24.31).9 Rarely, lesions have been described at other sites including the face, forearm, trunk, inguinal region, scrotum, buttocks, hallux, and nipple.9–12
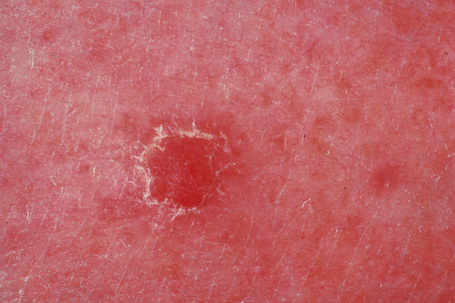
Fig. 24.31 Clear cell acanthoma: typical lesion on the shin of an elderly female.
By courtesy of the Institute of Dermatology, London, UK.
Occasionally, clear cell acanthoma presents as a polypoid lesion and large variants measuring up to 6 cm have been reported.11,13–18 Rare pigmented variants are also recognized.19 It often shows a collarette of scale, and erythematous puncta that bleed with minor trauma are commonly present on the surface.5 Clinically, it may resemble a pyogenic granuloma or an eccrine poroma. Individual case reports describe clear cell acanthoma arising within an epidermal nevus, in association with a melanocytic nevus, in a split-thickness skin graft and within a psoriatic plaque.17–22 Multiple lesions have been reported in association with ichthyosis.23
Pathogenesis and histological features
The precise nature of the clear cell acanthoma is unknown. Although variably regarded as an inflammatory epithelial hyperplasia, a hamartoma or a variant of a seborrheic keratosis, most authors believe it to be a benign neoplasm, although the cell of origin is subject to dispute.24 Derivation from epidermal, sebaceous, and sweat gland epithelium have all been proposed. The high glycogen content coupled with keratin and involucrin positivity and carcinoembryonic antigen (CEA) negativity has led some authors to suggest an origin from the follicular outer root sheath.24 However, considering the characteristic follicular sparing this seems an unlikely hypothesis. The presence of striking epithelial membrane antigen (EMA) expression makes an acrosyringeal derivation most improbable.25 Recently, the results of lectin binding suggest that it is of epidermal derivation.26 Furthermore, immunohistochemical studies on cytokeratin, involucrin, and filaggrin expression support an epidermal derivation and raise the possibility that clear cell acanthoma may represent an inflammatory dermatosis rather than a true neoplasm.27,28 This hypothesis is further supported by recent studies suggesting that, similar to psoriasis, keratinocyte growth factor (KGF) up-regulation may be responsible for keratinocyte hyperproliferation in clear cell acanthoma.29
It is composed of markedly acanthotic (often psoriasiform) epithelium, which has a characteristically clearly demarcated lateral border (Fig. 24.32). The epidermal ridges are commonly fused. Individual cells have clear cytoplasm due to the presence of abundant glycogen, best demonstrated with a periodic acid-Schiff (PAS) reaction (Fig. 24.33). Variably pigmented, dendritic melanocytes are sometimes present, both along the basal epithelial layer and also intermingled with keratinocytes in the upper layers of the lesion.19,30 This latter feature appears to be more common in patients of Mediterranean ancestry.31 In cases where pigmentation is clinically apparent, the confusing term clear cell melanoacanthoma (cf. seborrheic keratosis)32 or pigmented clear cell acanthoma has sometimes been applied.19,33 Typically, the intraepidermal portions of the adnexae are spared. Intralesional neutrophils are characteristic and are often evident within an overlying parakeratotic scale. The underlying dermal papillae commonly contain dilated capillaries, and an inflammatory cell infiltrate with a predominance of neutrophils is often present. These latter features show considerable overlap with psoriasis.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


