- Tumor suppressors are guardians against DNA damage induced, for example, by ultraviolet (UV) or irradiation (X-rays) or an excess of proliferative signals.
- Tumor suppressors that monitor DNA damage can help to repair the damage before cells divide. They can induce growth arrest at two DNA damage checkpoints: the first gap phase (G1) and the second gap phase (G2) of the cell cycle. This enables cells to check the integrity of the chromosomes before proceeding into DNA replication (S phase) or division (M phase). In the absence of these checkpoints, as a result of the loss of tumor suppressor function, the damage is not repaired and cells accumulate mutations that ultimately lead to cancer.
- Genetic (gene mutation or deletion) or epigenetic (promoter methylation, mutations in the promoter of genes) alterations in tumor suppressors such as RB, p53, or the genes that regulate them, are part of the life history of all cancer cells.
- The retinoblastoma protein, RB, and the transcription factor p53 control growth arrest, apoptosis, and differentiation, and their pathways collaborate in tumor prevention.
- RB binds to transcription factors to regulate cell proliferation. Its phosphorylation state affects its ability to bind these factors, E2Fs being the major ones. Phosphorylation of RB releases transcription factors that transcribe genes encoding proteins required for the initiation of DNA synthesis.
- Oncogenic and hyperproliferative stresses cause the activation of p53, which in turn restrains uncontrolled cell growth by blocking the cell cycle and inducing programmed cell death (apoptosis).
- RB and p53 are regulated by p16INK4a and p14ARF (p19Arf in mice).
Introduction
All that makes existence valuable to any one depends on the enforcement of restraints upon the actions of other people.
John Stuart Mill
Tumor suppressors are the cell’s guardians against DNA damage induced, for example, by ultraviolet (UV) exposure from sunlight, gamma irradiation (X-rays), chemotherapeutic drugs, or an excess of inappropriate proliferative signals. They prevent incipient cells from becoming malignant by arresting their proliferation or inducing them to commit suicide (apoptotic cell death). Tumor suppressors also monitor critical cellular checkpoints that govern the mitotic cycle, DNA repair, transcription, apoptosis, and differentiation. Some tumor suppressors prevent the inappropriate activation of signaling pathways involved in cell growth. The functional inactivation of tumor suppressors by mutation, deletion, or gene silencing creates an imbalance between proliferation, cell death, and differentiation programs that facilitates tumorigenesis. Some rare individuals are born with mutations in tumor suppressor genes that predispose them to develop cancer. Tumor suppressors, to date, represent about 0.1% (30) of the total number of genes (around 30 000) that make up the entire mammalian genome. This number is likely to increase as novel technologies are used to study cancer cells. It is now clear that cells have evolved tumor suppressors as part of multiple elaborate defense systems against unscheduled cellular proliferation.
As their name indicates, tumor suppressor proteins regulate cell proliferation by eliminating cells that are damaged (by cellular stresses such as UV, X-rays, chemotherapeutic drugs) or that abnormally proliferate in response to hyperproliferative signals. They are expressed by all mammalian cells and thus play a critical role in cancer prevention. Alfred G. Knudson and others, who proposed the “two-hits” hypothesis, introduced the concept of tumor suppression. Knudson theorized that the development of retinoblastoma, a rare cancer that occurs in the eyes of children, could be explained in familial cases by the acquisition from one parent (the first hit) of a mutated allele of a gene later called the retinoblastoma (RB) gene, and the occurrence of another mutation on the second allele (second hit) during adolescence. Nonfamilial or sporadic retinoblastoma, however, would occur as a result of mutations on both alleles in somatic cells, and in this case, the disease is not transmitted to the next generation. In each case, tumor development is recessive, since both copies of the gene must be inactivated for cancer to occur. This concept was later confirmed by studies on the genomic DNA from patients with retinoblastoma and other types of tumors. Since the discovery of RB, the first tumor suppressor identified, many tumor suppressor proteins have been characterized and shown to conform to the “two-hits” model proposed by Knudson.
The second tumor suppressor identified was the transcription factor p53. The realization that p53 was a tumor suppressor came from studies showing that enforced expression of wildtype p53, but not of mutant forms, induces a block in cell proliferation and transformation. This proliferation block is achieved, in part, by the activation of the growth arrest gene p21Cip1/Waf1, a cyclin-dependent kinase inhibitory protein, whose expression affects RB function. p53 was later found to be activated by DNA damage. The first link between p53 and the induction of apoptotic cell death, depending on the strength of the signals that activates it, was recognized in the early 1990s.
The RB and p53 tumor suppressor pathways are intertwined and mutations in genes in either pathway are likely events and even perhaps mandatory alterations in the life history of most tumor cells.
While these first two tumor suppressors continue to be the primary research interest of many cancer biologists, several other proteins have been found that share tumor-suppressing properties. Not surprisingly, many of these proteins participate in the regulation of cell proliferation and can be mapped to pathways that induce cell death (apoptosis) or growth arrest. In this chapter, we will discuss the function of several tumor suppressors that participate in the RB and p53 pathways, focusing on RB and p53, some of their upstream or downstream regulators, as well as tumor suppressors that regulate the flow of signals through critical growth stimulatory pathways. These tumor suppressors have been identified in many signaling pathways involved in multiple processes, including cell proliferation in response to receptor signals, DNA repair, transcriptional regulation, and cytoskeletal structure (see Table 7.1 and discussed in Chapters 4, 5, and 10).
Table 7.1 Tumor suppressors
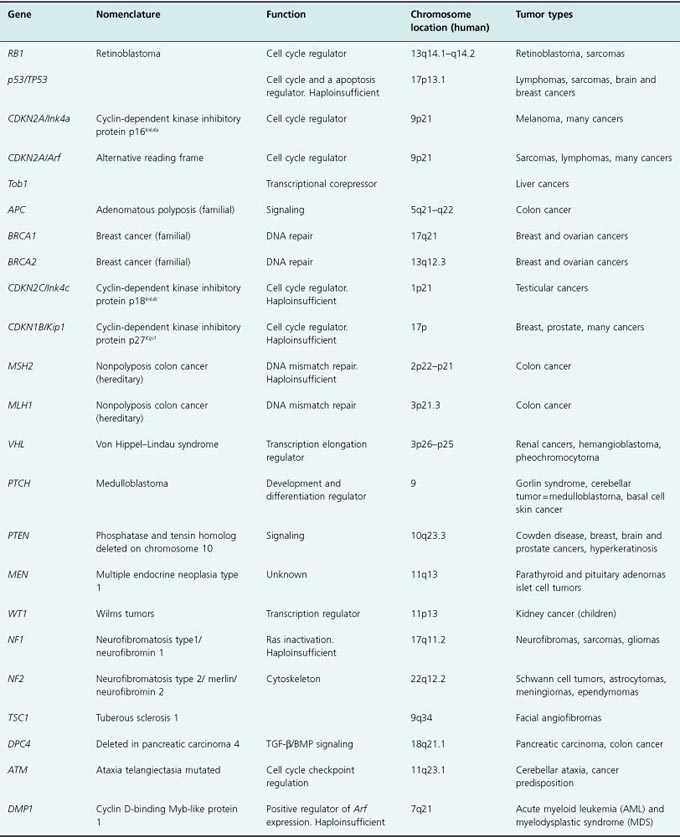
In addition to the common loss of both tumor suppressor alleles, tumor suppression can manifest itself in a haploinsufficient manner, meaning that loss of function of only one allele, or expression of only half of the amount of protein, is sufficient to sensitize cells to tumor formation. In these cases, the second allele remains intact and there is no loss of heterozygosity (also called LOH). Examples of haploinsufficient tumor suppressors include two cyclin-dependent kinase (CDK) inhibitory (CKI) proteins, p27Kip1 (CDKN1B in human) (Kip1, cdk inhibitor protein 1) and p18Ink4c (CDKN2C in human) (Ink4c, inhibitor of CDK4), reduced expression of which impairs the tumor suppressor activity of RB and p53, as well as PTEN, MSH2 (mismatch repair protein 2), and NF-1 (neurofibromatosis 1) (see Table 7.1, and discussed in Chapter 4).
Tumor suppressors have been conserved throughout evolution, and several have been found in species other than mammals, including frogs, fish, and flies. The focus of this chapter, however, will be the role of the major tumor suppressors in the RB and p53 pathways in preventing human cancer, as it relates to their structure and function in mammalian cells. All the concepts and principles attributed to RB and p53 also apply to other tumor suppressors. Indeed about 30 tumor suppressors are known, however, the vast majority of human cancers carry p53 and RB mutations or mutations in genes that regulate them.
The “Two-Hits” Hypothesis: Loss of Heterozygosity (LOH)
To lose one parent may be regarded as a misfortune; to lose both looks like carelessness.
Oscar Wilde
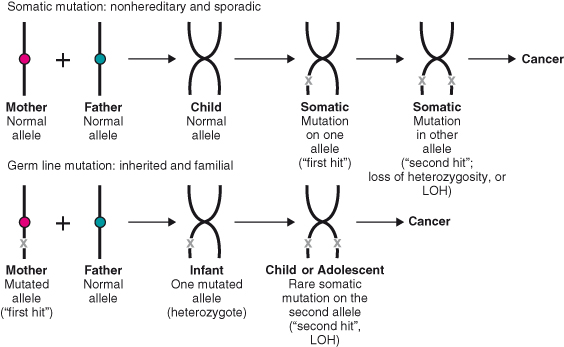
In 1971, Knudson advanced his “two-hits” hypothesis to explain how inherited (familial) cases of a rare cancer, called retinoblastoma, develop in the eyes of children (Fig. 7.1). Since then, this hypothesis has been confirmed in many cancers that occur as a result of the loss of a tumor suppressor. Knudson proposed that a child who inherits retinoblastoma acquires a mutated allele from one parent (the first hit) in a gene later called the retinoblastoma gene, RB. Heterozygosity for the RB allele is not sufficient to cause tumor formation and the child is initially asymptomatic. However, during early childhood, a second mutation or deletion on the other RB allele (second hit) occurs, inducing the loss of function of both alleles, loss of heterozygosity (LOH), in the retinal cells and the development of a tumor in this tissue. Nonfamilial or “sporadic” retinoblastoma also occurs as a result of mutations on both alleles (two hits) in retinal cells, but in this case, the disease is not transmitted to the next generation. Such mutations are termed “somatic mutations.” Regardless of the way the mutations occur, tumor development is recessive, since both copies of the gene must be inactivated for cancer to occur.
Figure 7.1 The “two-hits” hypothesis. Somatic mutation: In nonhereditary and sporadic mutations, genes inherited by both parents are normal, or wildtype. During early childhood or later, a mutation occurs in the allele of a gene, which represents the “first hit.” This is followed by the mutation or loss of the second allele of the same gene, the second hit. Loss of function of the second allele is called loss of heterozygosity or LOH. Germline mutation: In hereditary or familial cancers, one parent (in this example the mother) transmits a mutated gene. The child is born as heterozygote for that gene. During childhood – adolescence in retinoblastoma (RB) – a second mutation occurs in the second allele of the same gene, inducing LOH and cancer.
Mutation of RB genetically transmitted in the germline induces bilateral and multifocal retinal tumors, whereas sporadic retinoblastoma usually occurs in one eye only as a focal tumor. This concept was validated by studies on the genomic DNA from patients with retinoblastoma as well as DNA from patients with other cancers in which RB or other tumor suppressors were lost.
Many human tumors contain mutations in the RB gene, most of which occur in somatic cells, meaning that the mutation is not transmitted from one generation to the next.
Haploinsufficiency in Cancer
Haploinsufficiency is defined by the appearance of a phenotype in cells or an organism when only one of the two gene copies (also called alleles) is inactivated. For some tumor suppressor genes, the loss of a single allele is sufficient to induce susceptibility to tumor formation. In other words, these are haploinsufficient tumor suppressor genes. In this scenario, inactivation of one allele can be achieved by genetic (i.e. mutation or deletion) or epigenetic (i.e. transcriptional silencing by methylation, repressor complexes or mutations in the promoter region) events.
Epigenetic Events
Epigenetic regulation of genes can be achieved by methylation, repressor complexes, or mutations of the promoter regions or introns, thereby reducing or inducing a loss of messenger RNA transcription, and subsequent reduction or loss of protein expression. Methylation occurs on the cytosine nucleotide of CpG pairs, usually located in promoter regions. These pairs often cluster in gene regulatory regions referred to as CpG islands, (see Chapter 11). Methylation of CpG islands is associated with the formation of nuclease-resistant and compact chromatin structures, resulting in transcriptional silencing. Mutations in the promoter region can affect the efficiency with which transcription factors bind to specific DNA-binding domains. Transcriptional repression can be achieved by repressor protein complexes that bind to the promoter regions of affected genes.
Although the end result of genetic and epigenetic events is the same, that is, a given protein is not expressed, one event (epigenetic) involves the promoter or noncoding regions (introns) while the other (genetic) affects the coding regions of the gene. Epigenetic events are linked to cancer development and are equally important in the process of carcinogenesis. For example, expression of the tumor suppressor p16INK4a or p14ARF is often silenced by methylation of their promoters or by active transcriptional repression by repressor protein complexes that bind to their promoter rather than deletion or mutation of the genes (Box 7.1).
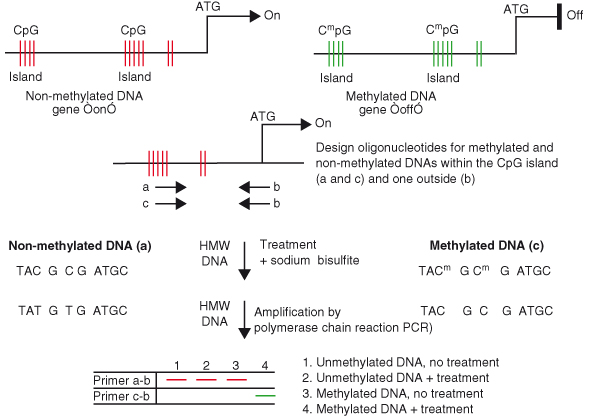
Thus, DNA methylation and gene silencing are linked to growth and differentiation regulatory pathways that are disrupted in nearly all cancer cells.
Definition of a Tumor Suppressor
Law; an ordinance of reason for the common good, made by him who has care of the community.
Thomas Aquinas
A tumor suppressor is a protein usually expressed in all cells at very low levels that slows or inhibits cell proliferation and prevents damaged or abnormal cells from becoming malignant if they have been exposed to DNA-damaging agents, such as ultraviolet (UV) or gamma-irradiation (X-rays), or to hyperproliferative signals induced by oncogenes (see Chapter 6). Enforced expression of tumor suppressors in normal cells invariably induces growth arrest or apoptosis, depending on the level of expression and the type of cell. Conversely, loss of tumor suppressor function sensitizes cells to tumor formation by creating a proliferative advantage that in turn usually leads to the accumulation of genetic alterations, and ultimately aggressive cancers. Of the ∼30 000 genes that make up the entire mammalian genome, tumor suppressors represent approximately 0.1%, although this number may increase as new technologies for studying tumor DNA become available and more tumor suppressors are identified (Table 7.1).
The Retinoblastoma Protein Family
Genomic Locus of RB: Mutations in Human Cancers
The retinoblastoma (RB) locus is located on human chromosome 13q14. It comprises 27 coding segments (exons) and spans a region of ∼200 kilobase pairs. Retinoblastoma can occur as a result of large deletions of coding exons or of more discrete mutations (e.g. point mutation) that induce either the loss of the messenger RNA or the expression of a nonfunctional RB protein. Most inactivating RB mutations lead to retinoblastoma, although less disruptive mutations, such as those in the promoter region of RB that affect its transcription, induce tumors less frequently.
RB is important in the control of proliferation and development of not only retinal cells, but also other tissues. Children with inherited retinoblastoma due to germline RB mutations are at higher risk of developing osteosarcomas later in adolescence because they lose the remaining normal RB allele. Certain sporadic human tumors with RB mutations that have been found at high frequency include carcinomas of the bladder (33%), breast (10%), and in particular small cell lung carcinomas (85%). Thus, the retinoblastoma protein RB plays a critical role in the control of neoplasia in a variety of tissues.
The RB Family of Proteins
RB is part of a family of three proteins that include p107 and p130. A unique gene located on a different chromosome encodes each family member. While the RB locus resides on the long arm of human chromosome 13 (13q14), p107 and p130 are located on human chromosomes 20q11.2 and 16q13, respectively. Unlike RB, these family members appear not to function as tumor suppressors, despite their close structural similarities to each other and to RB. Indeed, few mutations have been reported in the gene encoding p107 (a single B-cell lymphoma,) or p130 (in isolated lung tumors).
The retinoblastoma gene was the first tumor suppressor to be identified and cloned. The gene encodes a nuclear phosphoprotein of ∼110 kDa with distinct domains that are conserved between the other two family members, p107 and p130 (Fig. 7.2). The most conserved domain, termed the A–B “pocket,” comprises A and B boxes linked by a spacer region. While RB has little similarity to its family members outside the pocket domain, p107 and p130 share the highly conserved spacer domain. The spacer sequences are critical for the binding of cyclin A–CDK2 and cyclin E–CDK2 complexes. The A–B pocket is a structure rich in alpha helices to which numerous proteins are known to bind, including the E2F family of transcription factors and the cyclins. This pocket is also necessary for the binding of viral oncoproteins such as SV40 T antigen, adenovirus E1A, and E7 protein, all of which coopt RB to transform the cells in which their virus replicates. The binding of these viral proteins displaces the cellular proteins that normally interact with RB, inducing the loss of RB function.
Figure 7.2 Linear structures of the RB family of “pocket proteins.” Schematic representation of the three members of the RB family: pRb, p107, and p130. Note the conservation of many of the domains, including the A–B pocket, and the differences in the lengths of the spacer and the N- and C-termini.
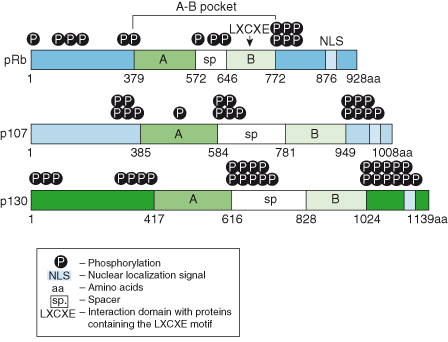
The recently solved crystal structure of the A–B pocket in complex with the oncoprotein E7 from the human papilloma virus (HPV) has revealed that the B domain binds to an “LXCXE motif” (where L is leucine, C is cysteine, E is glutamic acid, and X is any amino acid) in its binding partners. The A box determines and stabilizes the conformation of the B domain, and the A–B interface is also highly conserved. Therefore, mutations in either domain can disrupt the structure and function of RB. The C-terminal domain of RB proteins contains a nuclear localization signal (NLS) required for localization to the nucleus, and there are several phosphorylation sites in the N-terminus, spacer, and C domain downstream of the B box that affect RB protein function (see below).
RB and Its Binding Partners: Role During the Cell Cycle and Differentiation
The phosphorylation of RB fluctuates throughout the cell cycle (Box 7.2; see also Chapter 4). When cells are out of cycle in a state of quiescence (G0), or are in the early part of the first gap (G1) phase of the cell cycle, RB, p107, and p130 are underphosphorylated and in complex with other proteins, including the E2F transcription factors (E2F1–5) and their subunit partners, DP1 and DP2 (Fig. 7.3). While RB interacts principally with the “activating E2Fs,” E2F1, 2, and 3a, and prevents their transcriptional activity, it interacts to some extent with E2F4 and E2F5 to repress gene transcription. In contrast, p107 and p130 form complexes with only E2F4 and E2F5, and act as repressors of transcription in G0 and early G1 (Fig. 7.3).
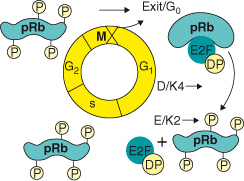
Figure 7.3 The RB family of proteins and G1 progression. In G0 and early G1, pRb, p107, and p130 are underphosphorylated and in complex with E2F4, E2F5, and the DP1 and DP2 proteins repress transcription. pRb also sequesters E2F1, 2, and 3a–DP complexes without contacting DNA. In G1, cyclin D–Cdk4/6 and cyclin E–Cdk2 holoenzymes phosphorylate pRb proteins, and relieve their repressive function, allowing E2Fs to induce the transcription of E2F-responsive genes required for S-phase entry.
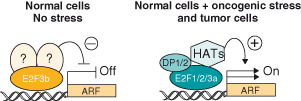
Two E2F3 proteins exist: E2F3a, the full-length protein that acts as an activating E2F, and E2F3b, a mutant form truncated at its N-terminus that represses transcription of proteins, including the tumor suppressor p19Arf (p14ARF in humans) (discussed in this chapter) by interacting with as yet unknown protein partners (Box 7.3). E2F6, another E2F family member, is part of a repressor polycomb complex that does not interact with the RB family of proteins. The most recently discovered E2F family member, E2F7, like E2F6, also acts as a transcriptional repressor and lacks an RB-binding domain.
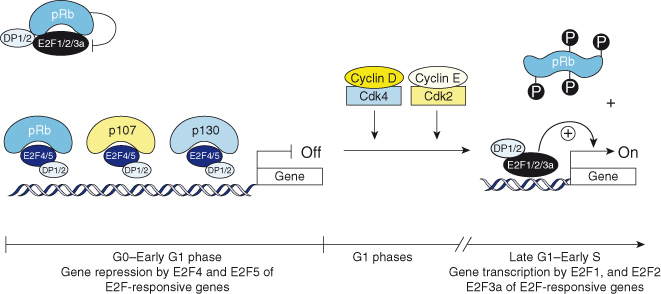
All E2Fs form complexes with the DP1 and DP2 subunits, which confer high-affinity DNA binding on the promoter of E2F-responsive genes. As cells progress through the G1 phase, RB becomes progressively phosphorylated on serine and threonine residues by cyclin–CDK holoenzymes that bind to RB via the LXCXE motif just described. Phosphorylation of RB is initiated by the holoenzyme cyclin D–CDK4, which forms in mid-G1 in response to mitogenic stimulation. This stimulation induces the transcription of D-type cyclins, which bind CDK4 and CDK6. RB becomes fully phosphorylated and inactivated by cyclin E–CDK2 holoenzymes. Phosphorylation requires the interaction of the holoenzymes with RB via the LXCXE motif located on the cyclins. Mutations in this motif prevent binding of the enzymatic complexes to RB and its phosphorylation. The phosphorylation of RB alters its conformation and growth-suppressive capabilities by causing the release of bound proteins, perhaps most importantly the E2F transcription factors. Freed from RB, the E2F1, 2, and 3a proteins and their DP partners activate the transcription of genes required for the cell to commit and proceed to the DNA synthetic (S) phase of the cell cycle (Fig. 7.3). RB is dephosphorylated in mitosis by the action of the phosphatase PP1alpha 2, so that the next mitotic cycle can begin.
Overexpression of wildtype but not mutated RB blocks cells in G1 by suppressing transcription and driving differentiation. RB, p130, and p107 interact with many proteins, but it is their binding to members of the E2F family of transcription factors that appears to be central to their role in governing DNA replication. As many as 110 proteins have been reported to interact with RB in vitro and in vivo, yet its ability to bind transcription factors, either repressing or stimulating their activity, appears to be the key to RB’s ability to suppress proliferation. Unphosphorylated RB family members and the proteins with which they interact actively repress gene expression by simultaneously recruiting histone deacetylases (HDACs), other remodeling factors, and E2F–DP1 to E2F-responsive promoters, and using the E2F–DP1 complex to position the complex onto specific promoters (Fig. 7.4).
Figure 7.4 The RB and E2F family of proteins: repression and activation of gene expression. Gene repression: (a) pRb, p107, and p130 repress transcription (OFF) by contacting DNA via E2F4, E2F5, and DP complexes and recruiting histone deacetylase (HDAC). Histones are not acetylated and the chromatin is condensed. (b) Repression can be mediated by E2F3b bound to as yet unidentified repressor proteins. (c) pRb sequesters E2F1, 2, and 3a and their DP partners and inhibits their function without contacting DNA. Gene activation: Phosphorylation of pRb releases E2F1, 2, and 3a, and DP partners, which recruit histone acetylase and induce the transcription of E2F-responsive genes. Histones are acetylated; the chromatin is opened, allowing gene transcription (ON).
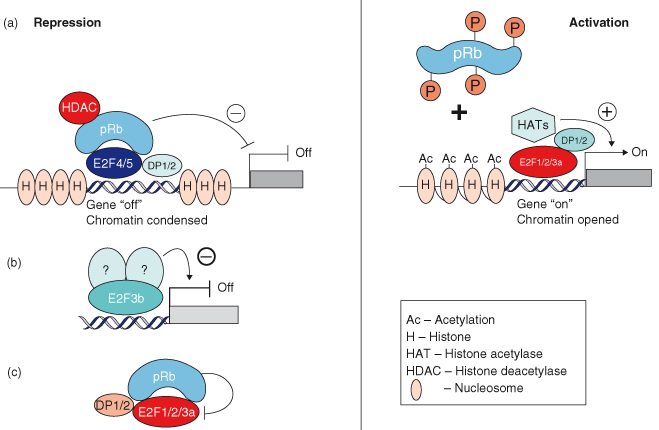
RB recruits HDACs to the B box of the pocket domain via the LXCXE motif on HDACs and in some cases by using a binding protein that bridges HDACs to RB, called RBP1 (RB-binding protein 1). By recruiting HDACs, RB not only forces cell cycle exit but also affects the expression of genes not involved in cell cycle regulation. p107 and p130 largely control the recruitment of HDACs to E2F-responsive promoters to repress their transcription, whereas RB is thought to bind to other transcription factors that regulate differentiation or senescence. In contrast, once RB is phosphorylated, gene activation proceeds by recruitment of histone acetylases to the activating E2F–DP complexes. This in turn induces the acetylation of histones, unwinding of the DNA that facilitates access to the DNA of the transcriptional machinery (Fig. 7.4).
The RB Signaling Pathway
The RB family of proteins negatively regulates cell proliferation by repressing the transcription of the genes responsible for progression through the G1 phase of the cell cycle and entry into S phase (Box 7.4). Relief of RB control is achieved by its phosphorylation by cyclin–CDK holoenzymes as discussed above. D-type cyclins are regulated by mitogenic signals via the Ras/Map kinase pathway that induces their transcription (Fig. 7.5). In that sense, D-type cyclins can be considered growth factor sensors. Once expressed, D-type cyclins bind to CDK4 and CDK6, a complex that is further activated by phosphorylation by a cyclin-dependent activating kinase, CAK.
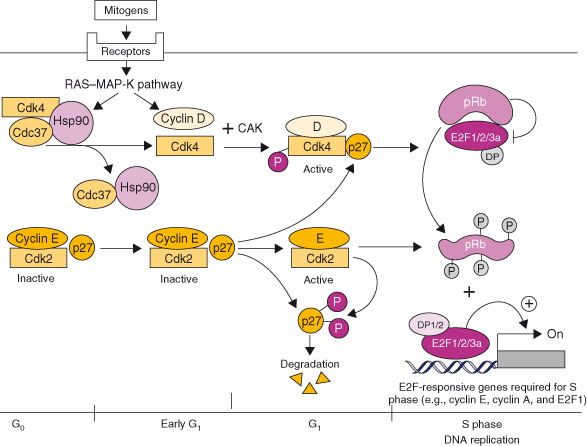
Figure 7.5 The Rb signaling pathway. In G0, Cdk4 is bound to the chaperone protein Hsp70, and Cdc37 while cyclin E–Cdk2 is bound to p27Kip1. Both kinases are inactive. In G1, upon mitogen activation, receptors activate the RAS–MAPK pathway that regulates cyclin D transcription and the release of Cdk4 from Hsp70 and Cdc37. Cdk4 kinase activity is induced by binding to D-type cyclins, its phosphorylation by the cyclin-dependent kinase (CAK) and p27Kip1 that serves as an assembly factor. The active cyclin D–Cdk4/6 holoenzyme initiates the phosphorylation of Rb. Binding of p27Kip1 to increased levels of cyclin D–Cdk4/6 complexes relieve the inhibition of cyclin E–Cdk2 complexes that complete the phosphorylation of Rb and phosphorylate p27Kip1, which is then degraded by the proteasome machinery. Phosphorylation of Rb releases E2F1, 2, and 3a that transactivate the expression of E2F-responsive genes, including cyclin E, cyclin A, and E2F1 and genes required for the initiation of DNA replication (S phase).
These holoenzymes are themselves regulated by inhibitory proteins called cyclin-dependent kinase inhibitory proteins (CKIs) that comprise two families: the INK4 family and the CIP/KIP family. INK4 proteins bind to and specifically negatively regulate the activity of the cyclin D-dependent kinases CDK4 and CDK6. The family consists of four members – p16INK4a, p15INK4b, p18INK4c, and p19INK4d – two of which, p16INK4a and p18INK4c, act as tumor suppressors. The CIP/KIP family has three members: p21CIP1/WAF1, a p53-responsive gene (see below), and p27KIP1 and p57KIP2, two tumor suppressors. All three members of this family are negative regulators of cyclin E– and cyclin A–CDK2 and of cyclin B–CDK1, while p27KIP1 and p21CIP1 also act as positive regulators of cyclin D–CDK4/6 by stimulating complex assembly. As cyclin D–CDK4 complexes accumulate in mid-G1, p27KIP1 and p21CIP1 are redistributed from cyclin E–CDK2 to cyclin D–CDK4/6 complexes, leading to the activation of cyclin E–CDK2, which completes the phosphorylation of RB, as described above. At this time, p27KIP1 itself is phosphorylated by active cyclin E–CDK2 and targeted for proteasomal degradation, which leads to the decrease in p27KIP1 levels that is required for S-phase entry (Fig. 7.6).
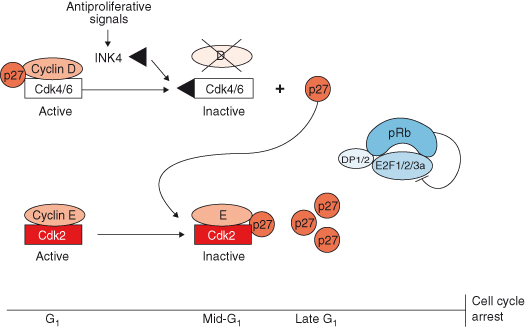
Figure 7.6 The cyclin-dependent kinase inhibitory (CKI) proteins from the Ink4 and Cip/Kip protein families collaborate to enforce cell cycle arrest. In mid-G1 and in response to antiproliferative signals, Ink4 proteins are induced. Ink4 proteins bind to Cdk4/6 and free cyclin D that is rapidly degraded. p27KIP1 is reassorted to cyclin E–Cdk2 complexes to inhibit their kinase activity. Rb phosphorylation is inhibited, cells can no longer enter S phase and cells are arrested in G1.
Cell-cycle exit, maintenance of quiescence or differentiated states, and senescence are achieved, in part, by the expression of the CKIs. CKI expression is induced by antiproliferative signals that include cell–cell interaction (see Chapter 12), or antimitotic signals, such as TGF-β (transforming growth factor) or the cytokine IL-10. Mitogens also upregulate INK4 proteins in mid-G1 of the normal cell cycle (Fig. 7.6; see also Chapter 5). As INK4 proteins accumulate, they bind with high affinity to the CDK4 and CDK6 moieties, thereby displacing the D-type cyclins from the complex, leading to their rapid degradation. Similarly, p27KIP1 and p21CIP1 reassort into the active cyclin E–CDK2 complexes and inhibit their activity. This prevents RB phosphorylation by both the cyclin D-containing and cyclin E–CDK holoenzymes and induces cell-cycle exit.
In addition to its role in regulating the cell mitotic cycle, the retinoblastoma protein participates in the DNA damage response (see Chapter 10), apoptosis (see Chapter 8), differentiation, and senescence (see Chapter 9).
RB and Human Cancers
There is now compelling evidence that several components of the RB pathway are mutated in human cancers, leading to the suggestion that the disabling of this pathway may be an inevitable event in the formation of most or all tumor cells. Components of this pathway act either as tumor suppressors that have been inactivated (e.g. INK4a, INK4c, KIP1, or RB), or as oncogenes that have been amplified (e.g. CDK4 or cyclin D1) (Table 7.2). While in cells containing a wildtype RB gene, the pRB protein represses the activating E2Fs via a sequestration mechanism in G0 and early G1 phase, loss of a functional RB protein unleashes the activating E2Fs that now transcribe genes at the inappropriate time, inducing either hyperproliferation or apoptosis (Box 7.5).
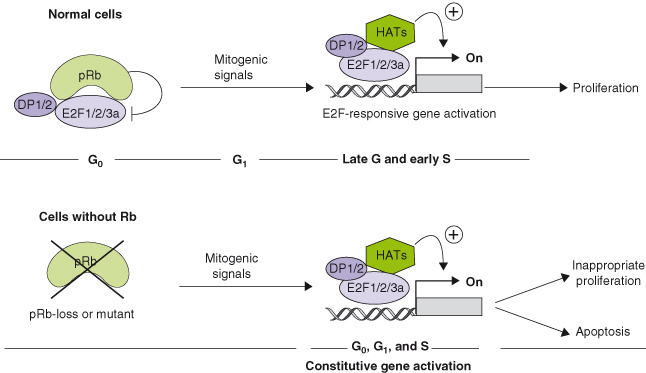
Table 7.2 The RB pathway in human cancers
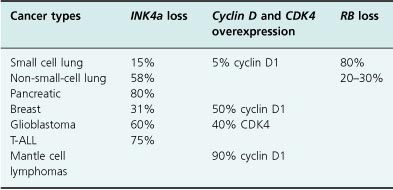
As discussed above, mutations in RB occur in several types of cancers, not just retinoblastoma. Similarly, while loss of p16INK4a functions by point mutations were initially discovered in familial melanoma, p16INK4a mutations, deletions, or epigenetic events have been found in many other cancers. Loss of the p18INK4c protein, presumably by epigenetic events affecting gene expression rather than the loss of the gene itself, is found in testicular cancers and correlates with the most aggressive forms of the disease. More recently, loss of p27KIP1 expression has been shown to correlate with a poor prognosis in not only colon and breast cancer, but in many other tumors, including prostate, bladder, lung, liver, ovary, stomach, and other organs.
RB function can also be abrogated by viral proteins while leaving the gene intact. The molecular mechanisms by which these viral proteins inactivate RB are still under intense investigation, although it is clear that RB disruption is required for both viral replication and transformation of mammalian cells. Indeed, small DNA tumor viruses, including human papilloma viruses (HPV) have evolved several mechanisms to inhibit the function of RB (discussed in Chapter 13). Women infected with HPV16 and HPV18 develop cervical cancers several years after the infection occurs. HPV16 encodes a protein (E7) that disrupts RB function by mimicking the binding of cyclin D1 to RB, thereby releasing E2Fs and possibly other proteins inhibited by pocket proteins. Indeed, RB/E2F complexes are absent in HPV-positive tumors and in cells expressing E7 alone.
E7 binds RB via its LXCXE motif. Very much like cyclin D1, mutations in the LXCXE domain prevent E7 from binding to RB, and block its ability to bypass RB-dependent cell-cycle arrest. By releasing E2Fs, the virus, which infects nondividing cells, forces induction of S phase, and essentially highjacks its host’s cell factors for its own replication. E7 also binds to p21Cip1, causing the release of proliferating cell nuclear antigen (PCNA), which also induces transcription of genes required for S-phase entry.
RB Family and Mouse Models
Mouse models in which RB and its family members are deleted have provided important information regarding the functions of the RB family in development and in adult tissues. Mice lacking two copies of Rb (Rb−/−) in the germline die during embryogenesis (E) between days 12.5 and 14.5 because of defects in red blood cell and skeletal differentiation, associated with apoptosis in the central nervous system (CNS) and liver (Table 7.3). More sophisticated genetic experiments were recently reported in which Rb was deleted conditionally or by using tetraploid aggregation. These experiments reveal an unexpected role for Rb in the placenta (i.e. outside the embryonic lineages) in the control of embryonic viability and development. They also demonstrate that the loss of Rb in the environment, rather than in the embryo proper, can affect CNS and red blood cell development.
Table 7.3 Mouse models of the Rb gene family
| Genotype | Phenotype |
| Rb−/− | Embryonic lethality (E12.5–14.5); CNS and liver apoptosis; erythropoiesis and skeletal defects |
| Rb tetraploid | Viable |
| Rb−/− (chimera) | Perinatal death; skeletal defects |
| Rb+/− | Neuroendocrine tumors by 12 months; pituitary adenocarcinomas (100%) and thyroid tumors with LOH |
| p107−/− | Tumor-free; viable |
| p130−/− | Tumor-free; strain-specific survival |
| p107−/−, p130−/− | Perinatal death and limb defects |
| Rb+/−, p107−/− | Retinal hyperplasia; pituitary and thyroid tumors with LOH for Rb |
| Rb−/− (chimera), p107−/− | Retinoblastomas; pituitary, and thyroid tumors |
| Rb+/−, p130−/− | No retinoblastomas or retinal defects; pituitary and thyroid tumors |
Mice heterozygotes for Rb function (Rb+/–) survive embryogenesis but are highly prone to the spontaneous development of neuroendocrine tumors of the pituitary and thyroid. Examination of these tumors invariably reveals the loss of the other Rb allele, which is analogous to the pattern of RB
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


